חלק א’: הדוגמה המרכזית של הגנטיקה
הגנום האנושי
- 95% מהגנום האנושי - רצפים לא מקודדים שתפקידם עדיין לא ברור לחלוטין
- כ־1% - מהגנים מקודדים חלבונים
- כמה אחוזים - אזורי בקרה שאנחנו קצת יותר מבינים
הדוגמה המרכזית (Central Dogma of Genetics)
הרעיון המרכזי שעליו בנויה היכולת של החיים על כדור הארץ:
\[\text{DNA} \xrightarrow{\text{Transcription}} \text{RNA} \xrightarrow{\text{Translation}} \text{Protein}\]- DNA צריך לעבור רפליקציה מדור לדור
- DNA עובר תעתוק (Transcription) ל־RNA
- RNA (השליח) מביא את המסר מהגרעין לציטופלזמה
- תרגום (Translation) של RNA לחלבון בריבוזומים
חלק ב’: רפליקציה של DNA (DNA Replication)
עקרונות יסוד
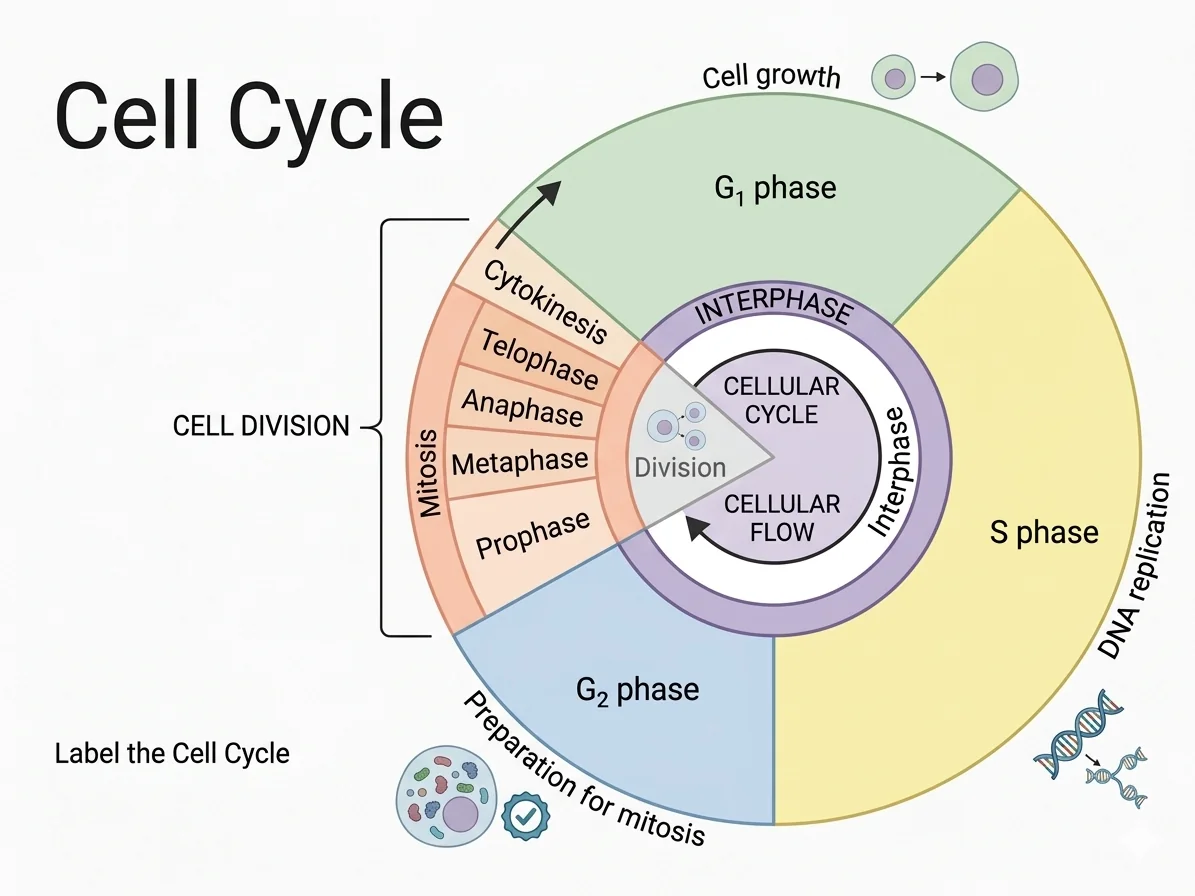
- רפליקציית ה־DNA מתרחשת בשלב $\text{S}$ של מחזור התא
- חייבת להיות מדויקת ביותר - טעויות יכולות לגרום:
- בתאים סומטיים: סרטן
- בתאי נבט (גמטות): מחלות גנטיות שעוברות לדור הבא
תהליך הרפליקציה
שלב ההתחלה (Initiation)
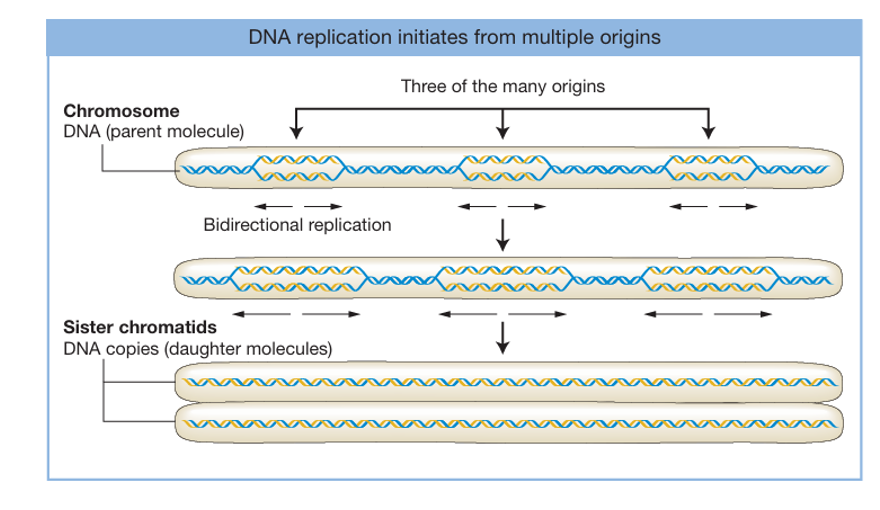
- Origin of Replication - נקודות התחלה באמצע הכרומוזומים (לא בקצוות)
- באאוקריוטים: אין רצף קבוע, תלוי בקונטקסט
- אזורים עשירים ב־A-T (קל יותר לפתוח - שני קשרי מימן בלבד)
- חלבונים מעורבים:
- Helicase - פותח את הסליל הכפול
- Gyrase/Topoisomerase - מונע התקשרויות וסיבובים לא רצויים
- SSB (Single Strand Binding proteins) - מחזיקים את הגדילים פתוחים
שלב ההתארכות (Elongation)
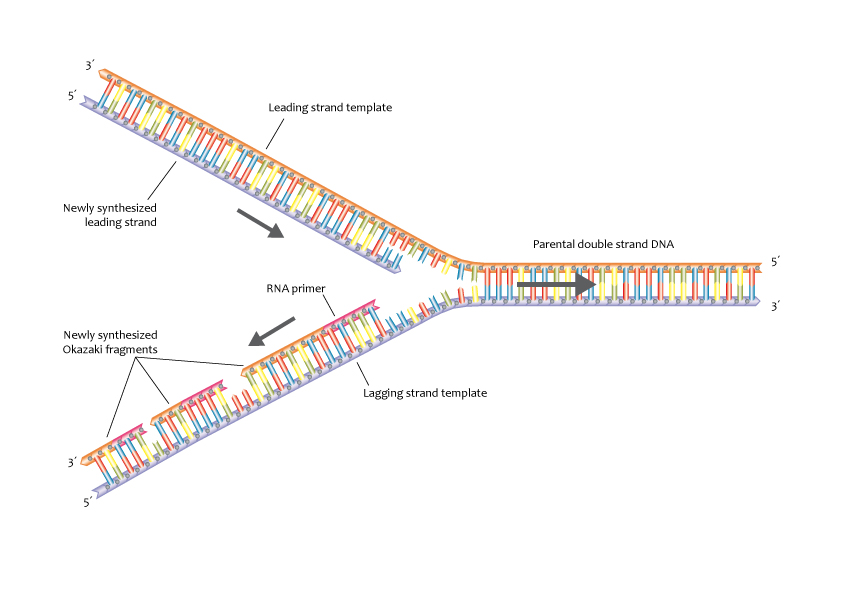
DNA Polymerase יכול לעבוד רק בכיוון 5’←3’, לכן:
- גדיל מוביל (Leading Strand)
- סינתזה רציפה בכיוון 5’←3’
- Primer אחד בהתחלה
- DNA Polymerase III מסנתז ברציפות
- גדיל מפגר (Lagging Strand)
- סינתזה לא רציפה במקטעים
- מקטעי אוקזקי (Okazaki Fragments) - קטעים קצרים
- Primase מוסיף primers רבים
- DNA Polymerase III מסנתז כל מקטע
- DNA Polymerase I מסיר primers וממלא פערים
- Ligase מחבר בין המקטעים
בעיית הטלומרים
- בקצות הכרומוזומים יש בעיה - אין איפה לשים primer
- טלומרים - רצפים חוזרים בקצוות
- טלומראז (Telomerase) - מאריך טלומרים
- קיצור טלומרים קשור להזדקנות
בקרת איכות ברפליקציה
- שכיחות טעות: $1$ ל־$10^7$ נוקלאוטידים
- מנגנוני תיקון:
- Proofreading על ידי DNA Polymerase
- DNA Repair mechanisms
- Checkpoints בסוף שלב S ולפני מיטוזה
- אם יש טעויות שלא ניתנות לתיקון ← אפופטוזיס
חלק ג’: תעתוק (Transcription)
הבדלים בין DNA ל־RNA
- RNA: חד־גדילי (Single-stranded)
- סוכר: ריבוז במקום דאוקסיריבוז
- בסיס: אורציל ($\ce{U}$) במקום תימין ($\ce{T}$)
- יכול לקבל מבנים מורכבים (loops, hairpins)
סוגי RNA
- mRNA (Messenger RNA) - נושא את המידע לסינתזת חלבונים
- rRNA (Ribosomal RNA) - מרכיב מבני בריבוזומים
- tRNA (Transfer RNA) - מעביר חומצות אמינו בתרגום
- Non-coding RNA - כולל microRNA, lncRNA ועוד - תפקידי בקרה
תהליך התעתוק
התחלה (Initiation)
- Promoter - אזור בקרה לפני הגן
- TATA Box - רצף TATAAA כ־25 בסיסים לפני תחילת התעתוק
- גורמי תעתוק (Transcription Factors)
- מזהים ונקשרים ל־promoter
- קוראים ל־RNA Polymerase
- אזורי בקרה נוספים:
- Enhancers - מגבירי ביטוי (יכולים להיות רחוקים)
- Silencers - משתיקי ביטוי
דוגמאות לבקרת תעתוק
-
ויטמין D כ־Transcription Factor:
- נקשר לרצפטור VDR (Vitamin D Receptor)
- הקומפלקס נכנס לגרעין
- נקשר לאזורים ספציפיים ב־DNA
- מפעיל גנים של ספיגת סידן
-
Lac Operon בחיידקים:
- ללא לקטוז: repressor חוסם את RNA polymerase
- עם לקטוז: repressor משתחרר, מתרחש תעתוק של אנזימי פירוק לקטוז
עיבוד RNA (RNA Processing)
שחבור (Splicing)
- הסרת אינטרונים (רצפים לא מקודדים)
- חיבור אקסונים (רצפים מקודדים)
- מוטציות באתרי שחבור יכולות לגרום למחלות
הוספות לקצוות
5' Cap- הגנה ויציבות3' Poly-A Tail- יציבות ותרגום
חלק ד’: תרגום (Translation)
הקוד הגנטי
- 64 קודונים (טריפלטים של 3 בסיסים)
- 20 חומצות אמינו
- קודון התחלה: AUG (מקודד למתיונין)
- קודוני סיום: UAA, UAG, UGA
- הקוד הוא מנוון (degenerate) - יותר מקודון אחד לחומצת אמינו
תהליך התרגום בריבוזום
מבנה הריבוזום
- שתי תת־יחידות (גדולה וקטנה)
- 3 אתרים:
- אתר A (Aminoacyl) - כניסת tRNA טעון
- אתר P (Peptidyl) - יצירת קשר פפטידי
- אתר E (Exit) - יציאת tRNA ריק
שלבי התרגום
- mRNA נכנס לריבוזום
- סריקה עד מציאת AUG
- tRNA עם מתיונין נקשר
- התקדמות קודון-קודון
- בקודון סיום - שחרור השרשרת
חלק ה’: יסודות הגנטיקה - חוקי מנדל
חוק ההפרדה השווה (Law of Equal Segregation)
- כל הורה תורם אלל אחד באופן אקראי
- נכון בכל המקרים
חוק הדומיננטיות (Law of Dominance)
- יש אללים דומיננטיים ורצסיביים
- נכון בחלק מהמקרים (לא תמיד)
חוק המבחר הבלתי תלוי (Independent Assortment)
לפי מנדל, כל תכונה עוברת בלי קשר לתכונה אחרת. כל גן עובר באופן רנדומאלי בלי קשר לגן לידו. זה לא מדויק כלל.
- לא נכון לגמרי - גנים על אותו כרומוזום עוברים ביחד
- Linkage - גנים קרובים על אותו כרומוזום
- Crossing Over במיוזה יוצר שונות נוספת. נותן
גנים שרחוקים מאוד לא בהכרח יעברו. זה תלוי במרחק הגנטי (לא תמיד פיזי) - linked vs unlinked.
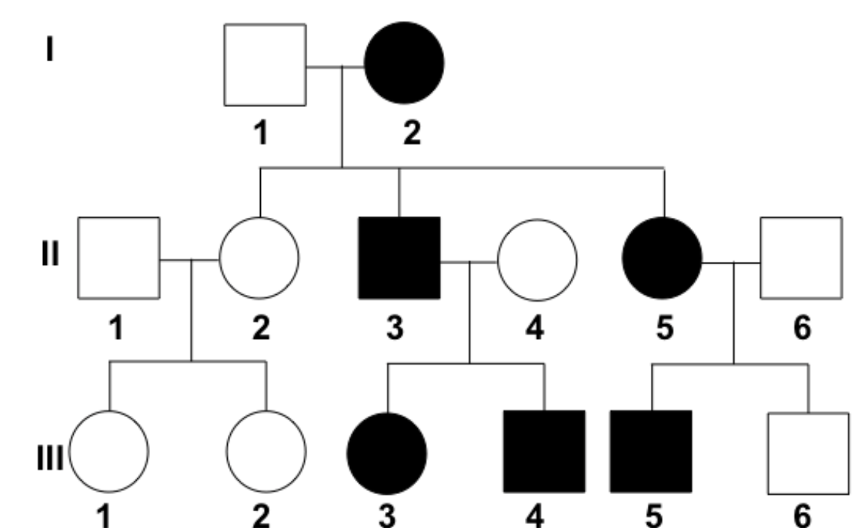
חלק ו’: מחלות גנטיות אוטוזומליות רצסיביות
עקרונות כלליים
- נדרשים שני אללים פגומים להופעת המחלה
- נשאים (הטרוזיגוטים) בדרך כלל בריאים
- שכיחות גבוהה יותר בנישואי קרובים
- עץ משפחה אופייני: הורים בריאים, ילדים חולים
□ ────┬───── ◐
│
┌───────┼───────┐
■ ○ □
◐ ────┬──── ◐
│
┌──────────┼──────────┬──────────┐
□ ■ ○ ●
□ ────┬──── ◐
│
┌──────────┼──────────┐
■ □ ◐ ──┬── □
│
┌──────────┼──────────┐
□ ■ ○
דוגמה 1: פנילקטונוריה (PKU)
Enzvmatic defects - not having enough of an enzyme to carry out a metabolic pathway.
פתופיזיולוגיה
- חסר באנזים Phenylalanine Hydroxylase
- פנילאלנין לא מתפרק לטירוזין
- הצטברות פנילאלנין ומטבוליטים רעילים
תסמינים
- פיגור שכלי (אם לא מטופל)
- עור בהיר (חסר מלנין)
- שיער בהיר
- עיניים בהירות
- פזילה
- היפרטוניות
- מיקרוצפליה
- ריח ״עובשי״ אופייני
טיפול
- דיאטה דלת פנילאלנין מלידה
- פורמולה מיוחדת עם חומצות אמינו (ללא פנילאלנין)
- הגבלה חמורה של חלבונים
- צריך כמות מינימלית של פנילאלנין (חומצת אמינו חיונית)
- המשך דיאטה לכל החיים (במיוחד בהריון)
אבחון
- סקר ילודים (Newborn Screening) - בדיקת דם מהעקב
- מדידת רמות פנילאלנין בדם
דוגמה 2: ציסטיק פיברוזיס (CF)
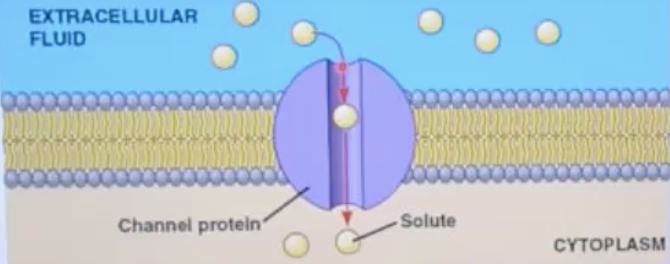
Channel Defect
גנטיקה
- גן:
CFTR(Cystic Fibrosis Transmembrane Regulator) - מוטציה שכיחה: ΔF508 (חסר של פנילאלנין במיקום 508)
- מעל 2000 מוטציות ידועות
- נשאות: 1:22 באוכלוסייה הכללית
פתופיזיולוגיה - סוגי מוטציות
- Class I-II: החלבון לא מגיע לממברנה
- Class III: החלבון מגיע אך לא נפתח
- Class IV: תעלה עובדת חלקית
- Class V: כמות מופחתת של חלבון תקין
תסמינים
ריאות:
- הפרשות סמיכות
- דלקות ריאה חוזרות
- ברונכיאקטזיות
לבלב:
- אי־ספיקה אקסוקרינית
- צואה שומנית
- אי־שגשוג
מעיים:
Meconium ileusבילודים (מקוניום סמיך)
מערכת רבייה:
- היעדר
vas deferensדו־צדדי בגברים (אי־פוריות)
טיפולים חדשניים (תלויי מוטציה)
- Potentiators - פותחים תעלות שמגיעות לממברנה
- Correctors - עוזרים לחלבון להתקפל ולהגיע לממברנה
- Amplifiers - מגבירים את ייצור החלבון
- אנטיביוטיקה (אמינוגליקוזידים) - עוקפים קודוני עצירה מוקדמים
חלק ז’: מחלות גנטיות אוטוזומליות דומיננטיות
מנגנונים למחלות דומיננטיות
1. Haploinsufficiency
- 50% מכמות החלבון לא מספיקה
- נפוץ בחלבונים מבניים
- דוגמה: Type I של Osteogenesis Imperfecta
2. Dominant Negative
- החלבון הפגום מפריע לחלבון התקין
- נפוץ בחלבונים המתארגנים במבנים מולטימריים
- דוגמה: Type II-III של Osteogenesis Imperfecta
3. Gain of Function
- החלבון רוכש פעילות חדשה או יתר
- דוגמה: אכונדרופלזיה
אוסטאוגנזיס אימפרפקטה (OI - ״מחלת העצמות השבירות״)
גנטיקה
- גנים:
COL1A1ו־COL1A2 - מקודדים לקולגן Type I
- מבנה: Triple helix של 3 שרשראות קולגן
- רצף חוזר:
Gly-X-Y(גליצין כל 3 חומצות אמינו)
סיווג וחומרה
Type I (קל) - Haploinsufficiency:
- שברים מתחילים בהליכה
- סקלרות כחולות
- אי־שגשוג קל
- חיים נורמליים יחסית
Type II (קטלני) - Dominant Negative:
- שברים תוך־רחמיים
- בית חזה קטן
- אי־ספיקה נשימתית
- מוות בינקות
Type III (חמור) - Dominant Negative:
- עיוותי עצמות חמורים
- קומה נמוכה מאוד
- שברים מרובים
- נכות קשה
תסמינים נוספים
- סקלרות כחולות - קולגן דליל בלובן העין
- Dentinogenesis Imperfecta - שיניים שבירות
- Wormian bones - עצמות נוספות בגולגולת
- ליקוי שמיעה
אכונדרופלזיה וגמדות (Achondroplasia)
גנטיקה
- גן:
FGFR3(Fibroblast Growth Factor Receptor 3) - מנגנון: Gain of Function
- 80% מוטציות De Novo
- Hot Spot - אותה מוטציה (G380R) ב־90% מהמקרים
- קשר לגיל אב מבוגר
פתופיזיולוגיה
- FGFR3 מעכב צמיחת עצם בלוחיות הגדילה
- המוטציה גורמת לעיכוב־יתר
- פגיעה בעיקר בעצמות ארוכות
תסמינים
- קומה נמוכה (גמדות)
- גפיים קצרות לא פרופורציונליות
- ראש גדול עם בליטת מצח
- גשר אף שקוע
- לורדוזיס מותנית
טיפול חדש
- תרופה מאושרת FDA מגיל 0
- פועלת downstream ב־pathway
- מנטרלת את העיכוב המוגבר
Thanatotropic Dysplasia
- מוטציה חמורה יותר ב־FGFR3
- בית חזה קטן מאוד
- אי־ספיקה נשימתית
- קטלני בדרך כלל
Homozygous Achondroplasia
- כאשר שני הורים עם אכונדרופלזיה מביאים ילד
- 25% סיכוי לילד עם 2 אללים פגומים
- פנוטיפ חמור הרבה יותר - בדרך כלל קטלני
Dravet Syndrome
- מחלת אפילפסיה חמורה בילדות
- דומיננטית אך בדרך כלל De Novo
- ילדים חולים בדרך כלל לא מביאים צאצאים
חלק ח’: מחלות עם הרחבת חזרות (Repeat Expansion Disorders)
מאפיינים כלליים
- רצפים חוזרים (בדרך כלל CAG) שמתרחבים
- Anticipation - החמרה מדור לדור:
- יותר חזרות
- גיל הופעה מוקדם יותר
- תסמינים חמורים יותר
מחלת הנטינגטון (Huntington Disease)
גנטיקה
- גן:
HTT - חזרות של רצף
CAGהמקודדות לגלוטמין (polyQ)
מספר חזרות וביטוי קליני
- 26-10: תקין
- 35-27: Pre-mutation (לא יציב)
- 40-36: Reduced penetrance
- מעל 40: מחלה מלאה
פתופיזיולוגיה
- עודף גלוטמינים גורם לקיפול שגוי של החלבון
- הצטברות חלבון במוח
- מוות של נוירונים
תסמינים של הנטינגטון
- תנועות כוריאיפורמיות
- דמנציה
- הפרעות פסיכיאטריות
- גיל הופעה: בדרך כלל 50-30 (תלוי במספר חזרות)
Anticipation
- סבא: 45 חזרות, תסמינים בגיל 60
- אב: 55 חזרות, תסמינים בגיל 40
- נכד: 70 חזרות, תסמינים בגיל 20
מחלות Fragile X
- הרחבת CGG ב־FMR1
- גורם לפיגור שכלי
- תורשה תלוית X עם אפקט anticipation
חלק ט’: מושגים חשובים נוספים
מוטציות De Novo
- מוטציות חדשות שלא עוברות בתורשה
- שכיחות גבוהה יותר:
- עם גיל האב המבוגר (תאי זרע מתחלקים כל הזמן)
- ב־Hot Spots (אזורים מועדים למוטציות)
- נפוצות במחלות דומיננטיות חמורות
Penetrance ו־Expressivity
Penetrance (חדירות)
- האם המוטציה תתבטא בכלל
- Complete (100%) או Incomplete (<100%)
Variable Expressivity
- חומרת הביטוי משתנה בין חולים
- אותה מוטציה - ספקטרום של תסמינים
מוזאיקה (Mosaicism)
Somatic Mosaicism
- המוטציה בחלק מתאי הגוף
- ביטוי קל יותר של המחלה
Germline Mosaicism
- המוטציה בחלק מתאי הנבט
- הורה בריא יכול להביא מספר ילדים חולים
- מסביר ״מוטציות דומיננטיות חוזרות״
חלק י’: חישוב סיכונים גנטיים
מחלות רצסיביות
הורים נשאים
- 25% ילד חולה
- 50% ילד נשא
- 25% ילד בריא לא נשא
- הסיכון זהה בכל הריון - ״זורקים את הקובייה מחדש״
חישוב סיכון עם נשאות באוכלוסייה
דוגמה - CF:
- נשאות באוכלוסייה: 1:22
- אם אחד ההורים נשא ידוע:
- סיכון שבן הזוג נשא: 1/22
- סיכון לילד חולה: 1/2 × 1/2 × 1/22 = 1/88
Obligate Carriers
- הורים לילד עם מחלה רצסיבית = בהכרח נשאים
- חריגים נדירים: מוזאיקות, מוטציה חדשה
חישוב נשאות באחים
לאחים בריאים של חולה במחלה רצסיבית:
- אם לא ידוע שבריאים: 50% סיכוי נשאות
- אם ידוע שבריאים (מחלת ילדות): 2/3 סיכוי נשאות
מחלות דומיננטיות
- הורה חולה: 50% סיכון לכל ילד
- שני הורים חולים: 75% ילד חולה (25% עלול להיות הומוזיגוט - חמור יותר)
חלק י״א: אבחון גנטי
סקר ילודים (Newborn Screening)
- בדיקת דם מהעקב
- מזהה מחלות מטבוליות (PKU, CF ועוד)
- מאפשר טיפול מוקדם
בדיקות טרום-לידתיות (Prenatal Tests)
- מי שפיר
- סיסי שלייה
- בדיקות גנטיות ממוקדות או רחבות
ייעוץ גנטי
- חישוב סיכונים
- הסבר על דפוסי תורשה
- אפשרויות לאבחון טרום-לידתי
- תמיכה פסיכולוגית
נקודות מפתח
- DNA Polymerase עובד רק 5’←3’ - קריטי להבנת רפליקציה
- מחלות רצסיביות - נשאות שכיחה, ביטוי רק בהומוזיגוטים
- מחלות דומיננטיות - מספיק אלל אחד פגום, לעתים De Novo
- Anticipation - החמרה מדור לדור במחלות חזרות
- הסיכון בכל הריון נפרד - לא תלוי בילדים קודמים
- Hot Spots - אזורים מועדים למוטציות
- טיפולים ממוקדי מוטציה - עתיד הרפואה הגנטית
שאלות תרגול מג׳ונרטות: מבנה DNA, רפליקציה, שעתוק ותרגום
- מבנה DNA - נוקלאוטידים, פורינים/פירימידינים, זיווג בסיסים (A-T 2H, C-G 3H), ארגון כרומטין (נוקלאוזומים ← כרומוזום)
- שכפול DNA - Helicase/Primase/Gyrase, Leading vs Lagging (Okazaki fragments, Ligase), טלומרים וטלומראז
- נקודות ביקורת - G2/M, Intra-S-phase
- RNA - הבדלים מ־DNA (Ribose, Uracil, חד־גדילי), סוגי RNA (mRNA, rRNA, tRNA)
- שעתוק - Initiation (Promoter, TATA box, TFs), Termination (AAUAAA), עיבוד pre-mRNA (capping, splicing/Lariat, Poly-A)
- תרגום - קוד גנטי, Anticodon
- מחלות רצסיביות - PKU (PAH, דיאטה, סריקת יילודים), CF (CFTR תעלת כלוריד, Ivacaftor, Modifiers)
- מנגנוני דומיננטיות - Haploinsufficiency vs Gain of Function vs Dominant Negative, OI קולגן
- Achondroplasia - FGFR3 Gain of Function, Hot spot, 80% de novo
- Anticipation - Trinucleotide repeats, Huntington
שאלה 1: מרכיבי הנוקלאוטיד
מהם שלושת המרכיבים של נוקלאוטיד ב־DNA?
- חומצת אמינו, סוכר ופוספט
- בסיס חנקני, סוכר ריבוז וקבוצת פוספט
- בסיס חנקני, סוכר דאוקסיריבוז וקבוצת פוספט
- בסיס חנקני, סוכר דאוקסיריבוז ושומן
פתרון
התשובה הנכונה היא (3).
מהשקף (שקף 4): נוקלאוטיד = Base + Sugar (Deoxyribose) + Phosphate.
הנוקלאוטידים מחוברים זה לזה דרך קשרי Phosphodiester בין הסוכרים. סיבוב אחד של הסליל הכפול = 10 זוגות בסיסים (bp).
תשובה (2) שגויה כי ב־DNA הסוכר הוא Deoxyribose (חסר OH בעמדה 2’), לא Ribose - הסוכר Ribose מאפיין את RNA.
שאלה 2: פורינים ופירימידינים
אילו בסיסים חנקניים הם פורינים ואילו פירימידינים?
- פורינים: A, C; פירימידינים: G, T
- פורינים: A, T; פירימידינים: C, G
- פורינים: A, G; פירימידינים: C, T
- פורינים: C, G; פירימידינים: A, T
פתרון
התשובה הנכונה היא (3).
מהשקף (שקף 5):
פורינים (Purines) - טבעת כפולה: Adenine (A) ו־Guanine (G).
פירימידינים (Pyrimidines) - טבעת בודדת: Cytosine (C) ו־Thymine (T).
כלל זיכרון: PYrimidines = C, T, U (אותיות “חדות” - Y). PURines = A, G (אותיות “עגולות”).
שימו לב: ב־RNA, Uracil (U) מחליף את Thymine (T) - שניהם פירימידינים.
שאלה 3: זיווג בסיסים משלימים
מהו הזיווג הנכון בין בסיסי DNA ואיזה קשר חזק יותר?
- A-G ו־C-T; הקשר A-G חזק יותר
- A-T ו־C-G; הקשר A-T חזק יותר כי יש בו 3 קשרי מימן
- A-T ו־C-G; הקשר C-G חזק יותר כי יש בו 3 קשרי מימן
- A-C ו־G-T; הקשר G-T חזק יותר
פתרון
התשובה הנכונה היא (3).
מהשקף (שקפים 6-7):
A pairs with T - 2 קשרי מימן. C pairs with G - 3 קשרי מימן ← קשר חזק יותר.
“C to G is a stronger bond”
משמעות מעשית: DNA עשיר ב־GC יהיה יציב יותר ודורש טמפרטורה גבוהה יותר לדנטורציה (הפרדת הגדילים). זה רלוונטי ב־PCR ובמחקר גנטי.
הגדילים רצים בכיוונים הפוכים: 5’←3’ מול 3’←5’ (אנטי-פרלליים).
שאלה 4: ארגון הכרומטין
מהי יחידת הארגון הבסיסית של הכרומטין?
- כרומוזום שלם עם צנטרומר וטלומרים
- נוקלאוזום - DNA כרוך סביב חלבוני היסטון
- סיב כרומטין מעובה בעובי 300 ננומטר
- גדיל DNA כפול חשוף ללא חלבונים
פתרון
התשובה הנכונה היא (2).
מהשקף (שקף 8): הדיאגרמה מראה את רמות האריזה מ־DNA חשוף ועד כרומוזום מלא:
- DNA double helix (2 nm)
- “Beads on a string” - נוקלאוזומים (11 nm)
- סיב כרומטין של 30 nm
- קטע כרומוזום פתוח (300 nm)
- קטע מעובה (700 nm)
- כרומוזום מיטוטי שלם (1,400 nm)
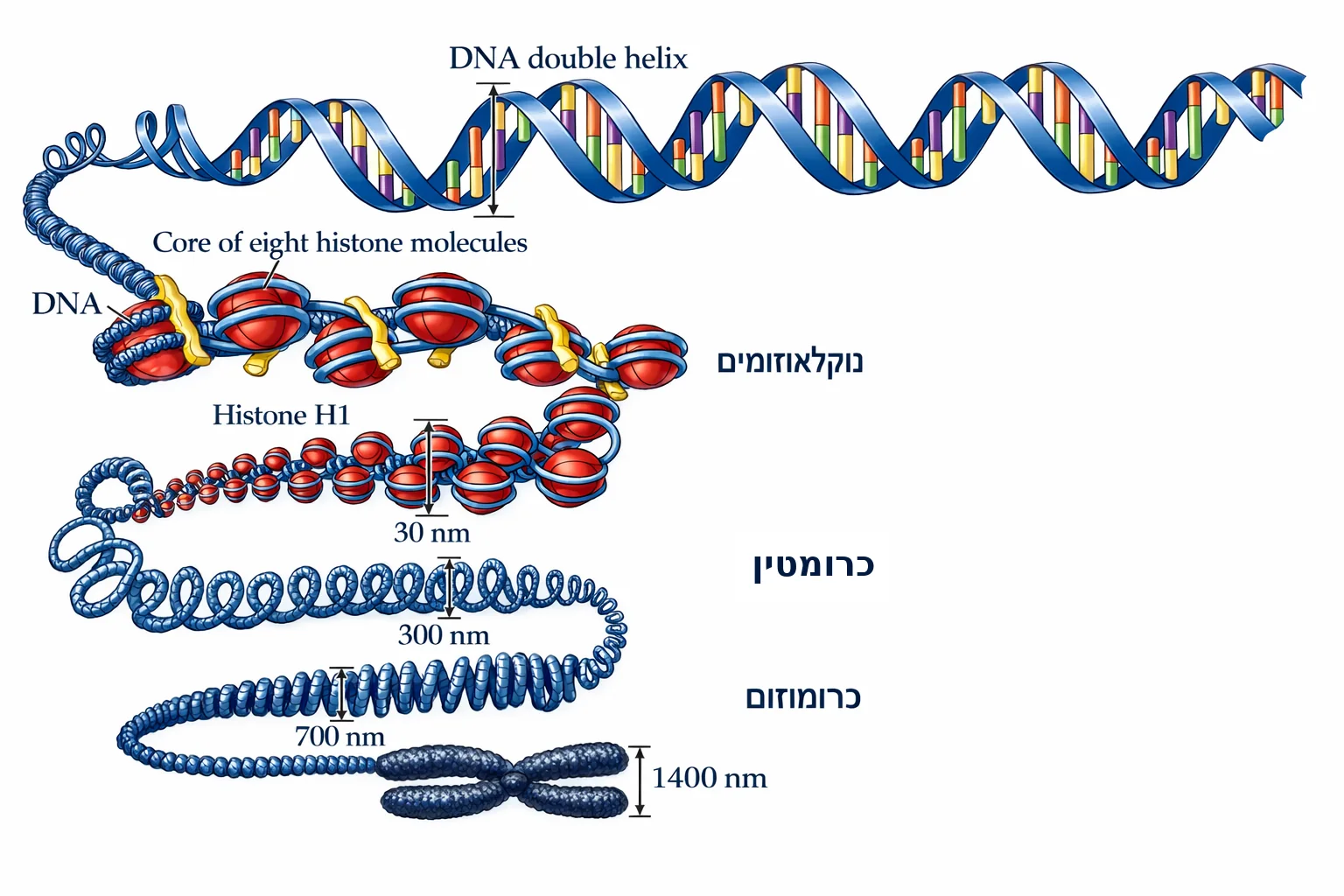
הנוקלאוזום = DNA כרוך סביב אוקטמר של היסטונים (8 חלבונים). אותו שקף גם מציג את מבנה הגן: Promoter ← Exons/Introns ← poly A.
שאלה 5: שלבי שכפול DNA - Initiation
מה תפקידם של Helicase ו־Primase בשלב ה־Initiation?
- Helicase מעתיקה את ה־DNA ו־Primase מחברת קטעים
- Helicase פותחת את הסליל הכפול ו־Primase מניחה פריימר RNA
- Helicase מתקנת שגיאות ו־Primase מוחקת אותן
- Helicase מוסיפה טלומרים ו־Primase מקצרת אותם
פתרון
התשובה הנכונה היא (2).
מהשקפים (שקפים 12, 15):
Initiation:
- Helicase - פותחת (unwinds) את הסליל הכפול ב־Origin of Replication.
- Gyrase (Topoisomerase) - מסירה את המתח (supercoiling) שנוצר מהפתיחה.
- Primase - מניחה פריימר RNA קצר על תבנית ה־DNA, כי DNA Polymerase לא יכולה להתחיל סינתזה מאפס.
- SSB (Single Strand Binding proteins) - מייצבות את הגדילים הפתוחים.
שאלה 6: Leading strand מול Lagging strand
מה ההבדל בין ה־Leading strand ל־Lagging strand בשכפול DNA?
- שניהם נבנים ברציפות אבל בכיוונים הפוכים
- ה־Leading נבנה ברציפות וה־Lagging בקטעי אוקזקי שמחוברים ע”י Ligase
- ה־Lagging נבנה ברציפות וה־Leading בקטעי אוקזקי שמחוברים ע”י Ligase
- ה־Leading נבנה ע”י RNA Polymerase וה־Lagging ע”י DNA Polymerase
פתרון
התשובה הנכונה היא (2).
מהשקפים (שקפים 13, 15):
DNA Polymerase III יכולה לבנות רק בכיוון 5’←3’.
Leading strand: התבנית רצה בכיוון 3’←5’ ← הגדיל החדש נבנה ברציפות (Continuous synthesis) לכיוון מזלג השכפול.
Lagging strand: התבנית רצה בכיוון 5’←3’ ← הגדיל החדש נבנה בקטעים קצרים הנקראים Okazaki fragments, כל אחד מתחיל עם RNA primer. DNA Polymerase I מסירה את הפריימרים וממלאת את הפערים. DNA Ligase מחברת את הקטעים.
שאלה 7: טלומרים וטלומראז
מדוע הכרומוזומים מתקצרים בכל סבב שכפול ומה תפקיד הטלומראז?
- כי DNA Polymerase מוחקת את הקצוות בכוונה; טלומראז מתקן שגיאות
- כי לא ניתן לשכפל את קצה ה־Lagging strand לאחר הסרת הפריימר; טלומראז מאריך טלומרים
- כי Helicase חותכת את הקצוות; טלומראז מדביק אותם
- כי הקצוות נהרסים ע”י אנזימי פירוק; טלומראז מגן עליהם כימית
פתרון
התשובה הנכונה היא (2).
מהשקף (שקף 14):
בעיית קצה השכפול (End Replication Problem): לאחר הסרת RNA primer מקצה ה־Lagging strand, DNA Polymerase לא יכולה למלא את הפער כי אין פריימר שמשמש כנקודת התחלה ← הכרומוזום מתקצר.
Telomerase - אנזים מסוג reverse transcriptase שמאריך את רצף הטלומר. פעיל בעיקר בתאי נבט ובתאי גזע. רוב התאים הסומטיים חסרים טלומראז ← קיצור הדרגתי ← הזדקנות תאית. הפעלה מחדש של טלומראז בתאים סרטניים ← התחלקות בלתי מוגבלת.
שאלה 8: נקודות ביקורת במחזור התא
מהו תפקיד ה־G2/M Checkpoint?
- לוודא שהתא גדל מספיק לפני שכפול DNA
- לוודא שהכרומוזומים שוכפלו במדויק לפני כניסה למיוזה
- לוודא שהכרומוזומים שוכפלו במדויק לפני כניסה למיטוזה
- לקבוע אם התא יעבור אפופטוזיס לאחר המיטוזה
פתרון
התשובה הנכונה היא (3).
מהשקף (שקף 17):
G2/M Checkpoint - ממוקם במעבר בין G2 ל־Mitosis:
“Ensures that all chromosomes have been accurately and completely replicated without any damage before the cell enters mitosis (M phase).”
נקודות ביקורת נוספות:
- G1 Checkpoint (Restriction) - בודק אם התא מוכן לשכפול DNA.
- Intra-S-phase Checkpoint - עוקב אחר התקדמות שכפול DNA, מייצב מזלגות שכפול שנעצרו, ומונע ירי של Origins חדשים אם יש נזק.
- M Checkpoint - בודק שכל הכרומוזומים מחוברים לציר.
שאלה 9: הבדלים בין RNA ל־DNA
אילו שלושה הבדלים מרכזיים קיימים בין RNA ל־DNA?
- RNA דו־גדילי, מכיל Thymine ו־Deoxyribose במקום Ribose
- RNA חד־גדילי, מכיל Uracil במקום Thymine ו־Ribose במקום Deoxyribose
- RNA חד־גדילי, מכיל Thymine במקום Uracil ו־Ribose במקום Deoxyribose
- RNA דו־גדילי, מכיל Uracil וסוכר Ribose במקום Deoxyribose
פתרון
התשובה הנכונה היא (2).
מהשקף (שקף 20):
שלושה הבדלים מרכזיים:
- RNA = Single strand (חד־גדילי) ≠ DNA = Double strand
- RNA = Ribose sugar (עם OH בעמדה 2’) ≠ DNA = Deoxyribose (H בעמדה 2’)
- RNA = Uracil (U) ≠ DNA = Thymine (T)
ה־OH בעמדה 2’ של Ribose מאפשר ל־RNA ליצור מבנים תלת־ממדיים מורכבים. U יכול להיקשר גם ל־A וגם ל־G ב־RNA (Wobble pairing) - מה שמאפשר מבנים מורכבים.
שאלה 10: סוגי RNA
מהם שלושת סוגי ה־RNA העיקריים ומה תפקידם?
- mRNA מתרגם חלבונים; rRNA נושא חומצות אמינו; tRNA בונה ריבוזומים
- mRNA נושא מידע גנטי; rRNA רכיב מבני בריבוזום; tRNA מביא חומצות אמינו
- mRNA מתקן DNA; rRNA מעתיק DNA; tRNA מפרק חלבונים
- mRNA בונה ריבוזומים; rRNA נושא מידע גנטי; tRNA נושא חומצות אמינו
פתרון
התשובה הנכונה היא (2).
מהשקף (שקף 22):
mRNA (Messenger RNA) - נושא את המידע הגנטי מה־DNA לריבוזום. מהווה “תבנית” לתרגום.
rRNA (Ribosomal RNA) - רכיב מבני וקטליטי של הריבוזום. הרכיב העיקרי של יחידות המשנה של הריבוזום.
tRNA (Transfer RNA) - מביא חומצות אמינו לריבוזום. בעל מבנה “תלתן” עם Anticodon בצד אחד וחומצת אמינו בצד השני.
שאלה 11: שלבי השעתוק - Initiation
מה קורה בשלב ה־Initiation של השעתוק?
- הריבוזום נקשר ל־mRNA ומתחיל לתרגם חלבון
- RNA Polymerase נקשר ל־Promoter ומתחיל לבנות RNA
- ה־DNA נפתח ונוצרים מקטעי אוקזקי חדשים
- ה־mRNA עובר שחבור (splicing) לפני היציאה מהגרעין
פתרון
התשובה הנכונה היא (2).
מהשקפים (שקפים 26-25, 30):
Initiation:
- ה־DNA נפתח (Unwind).
- רק גדיל אחד משמש כ־Template.
- RNA Polymerase נקשר ל־Promoter - רצף DNA לפני אתר תחילת השעתוק.
- ה־Promoter מכיל TATA box - רצף שמזוהה ע”י RNA Polymerase.
- Transcription Factors - חלבונים שנקשרים לרצפים מסביב לגן ומשפיעים על השעתוק (מפעילים או מעכבים).
“Transcription determines gene expression so needs to be highly regulated”
שאלה 12: Elongation ו־Termination של שעתוק
כיצד מסתיים תהליך השעתוק?
- RNA Polymerase מגיעה לקצה הכרומוזום ונופלת
- RNA Polymerase מגיעה לסיגנל הפולי-אדנילציה (
5'AAUAAA3') - הריבוזום מפסיק את RNA Polymerase ומשחרר אותה
- DNA Ligase חותכת את ה־RNA ומשחררת אותו
פתרון
התשובה הנכונה היא (2).
מהשקפים (שקפים 30-29):
Elongation: ממשיך מאתר ההתחלה, כולל 5’UTR, אקסונים ואינטרונים, קודון עצירה ו־3’UTR.
Termination: “RNA polymerase reaches the polyadenylation signal (5’AAUAAA3’)”
← אינטראקציה בין חלבונים ל־Polymerase ← הוספת Poly-A tail ← שחרור.
שלוש עיבודים ל־pre-mRNA:
- 5’ Capping - הוספת כיפה בקצה 5’
- Splicing - הסרת אינטרונים
- Polyadenylation - הוספת זנב Poly-A בקצה 3’
שאלה 13: עיבוד pre-mRNA - שחבור (Splicing)
מה קורה בתהליך ה־Splicing?
- ה־mRNA מתחלק לכמה עותקים זהים וכל אחד עובר עיבוד שונה
- האינטרונים מוסרים והאקסונים מחוברים יחד ליצירת mRNA בוגר
- האקסונים מוסרים והאינטרונים נשארים ב־mRNA הבוגר
- ה־DNA עצמו עובר תיקון ומוטציות מוסרות
פתרון
התשובה הנכונה היא (2).
מהשקפים (שקפים 34-35):
שחבור (Splicing):
- pre-mRNA מכיל אקסונים (Exons = EXpressed) ואינטרונים (Introns = INtervening).
- האינטרונים מוסרים - נוצר מבנה Lariat (לולאה) שמפורק.
- האקסונים מחוברים (Linked exons) ← mRNA בוגר.
מבנה הגן (שקף 35): DNA ← Promoter ← [Potential regulatory elements] ← Transcription start site ← Exon-Intron-Exon-Intron-Exon ← Transcription stop site.
חשוב: mRNA בוגר = רק אקסונים (+ 5’UTR + 3’UTR).
שאלה 14: הקוד הגנטי ו־tRNA
כמה נוקלאוטידים מקודדים חומצת אמינו אחת ומהו תפקיד ה־Anticodon?
- 2 נוקלאוטידים; ה־Anticodon על ה־mRNA נקשר לקודון
- 3 נוקלאוטידים; ה־Anticodon על ה־tRNA נקשר לקודון
- 4 נוקלאוטידים; ה־Anticodon על ה־rRNA נקשר לקודון
- 1 נוקלאוטיד; ה־Anticodon זהה לקודון על ה־mRNA
פתרון
התשובה הנכונה היא (2).
מהשקפים (שקפים 37-38):
הקוד הגנטי: כל 3 נוקלאוטידים (Codon) מקודדים חומצת אמינו אחת. סה”כ 4³ = 64 קודונים, מתוכם 61 מקודדים חומצות אמינו ו־3 הם קודוני Stop (UAA, UAG, UGA).
הקוד דגנרטיבי (Degenerate) - חומצות אמינו רבות מקודדות ע”י יותר מקודון אחד.
tRNA: בצד אחד - Anticodon (משלים לקודון על ה־mRNA). בצד השני - חומצת אמינו ספציפית. ה־tRNA הוא “המתרגם” בין שפת הנוקלאוטידים לשפת החלבונים.
שאלה 15: PKU - פנילקטונוריה
מהו הפגם ב־PKU ומהו הטיפול?
- חסר באנזים שמפרק גלוקוז; דיאטה דלת סוכר
- חסר באנזים PAH שממיר Phe ל־Tyr; דיאטה דלת פנילאלנין
- עודף באנזים שמייצר טירוזין; תוספת פנילאלנין
- חסר בקולטן ממברנלי לפנילאלנין; החלפת קולטנים
פתרון
התשובה הנכונה היא (2).
מהשקפים (שקפים 54-50, 57):
PKU (Phenylketonuria): מחלה אוטוזומלית רצסיבית.
האנזים Phenylalanine Hydroxylase (PAH) אינו פעיל ← Phenylalanine מצטבר ← נזק מוחי.
סימפטומים: פיגור שכלי חמור, פרכוסים, מיקרוצפליה, ריח עובש מהגוף, אקזמה.
טיפול: דיאטה דלת Phenylalanine (תחליפי חלב מיוחדים). תוספת BH4 (Tetrahydrobiopterin) - קופקטור לאנזים.
סריקת יילודים (Newborn Screening) - בדיקת דם מיד לאחר הלידה מאפשרת התחלת טיפול מוקדם ומניעת נזק מוחי.
שאלה 16: Cystic Fibrosis - פגם בתעלה
מהו סוג הפגם ב־Cystic Fibrosis?
- פגם אנזימטי - האנזים לא מפרק מוקוס
- פגם בקולטן - הקולטן לא נקשר לליגנד שלו
- פגם בתעלת כלוריד - תעלת יונים לא מתפקדת
- פגם בחלבון מבני - הקולגן לא נוצר כראוי
פתרון
התשובה הנכונה היא (3).
מהשקפים (שקפים 58, 60-63):
CF (Cystic Fibrosis): מחלה אוטוזומלית רצסיבית. פגם בגן CFTR - מקודד לתעלת כלוריד (Channel Defect).
התעלה לא מתפקדת ← הפרשת מוקוס עבה ודביק ← פגיעה בריאות, לבלב, מערכת עיכול.
סוגי מוטציות שונות בגן CFTR - בין השאר מוטציות שפוגעות בייצור החלבון, בתפקודו או במיקומו בממברנה. טיפולים כמו Ivacaftor מכוונים לסוגי מוטציות ספציפיים.
Modifiers (שקף 64): חומרת המחלה מושפעת ע”י גנים מווסתים (Modifier genes) וגורמי סביבה.
שאלה 17: מנגנוני דומיננטיות - שלושה סוגים
מהם שלושת המנגנונים להורשה דומיננטית?
- Frameshift, Nonsense, Missense
- Haploinsufficiency, Gain of Function, Dominant Negative
- Deletion, Insertion, Duplication
- Epistasis, Codominance, Incomplete Dominance
פתרון
התשובה הנכונה היא (2).
מהשקף (שקף 69):
שלושת המנגנונים להורשה דומיננטית:
-
Haploinsufficiency - עותק אחד תקין לא מספיק. התא צריך 100% תוצר, מקבל רק 50% ← מחלה. דוגמה: OI קל (Type I) - חסר בכמות קולגן.
-
Gain of Function - המוטציה יוצרת חלבון עם פעילות חדשה או מוגברת. דוגמה: Achondroplasia - FGFR3 פעיל תמיד (שקף 82).
-
Dominant Negative - החלבון המוטנטי “מרעיל” את התוצר התקין. דוגמה: OI חמור - שרשרת קולגן פגומה הורסת את ה־Triple helix (שקפים 76-77).
שאלה 18: OI - Dominant Negative מול Haploinsufficiency
מדוע OI Type I (Haploinsufficiency) קל יותר מ־OI חמור (Dominant Negative)?
- ב־Type I יש יותר מוטציות מאשר בחמור
- ב־Type I נוצר פחות קולגן אך הוא תקין; ב־DN הקולגן הפגום הורס גם את התקין
- ב־Type I אין קולגן בכלל; ב־DN יש קולגן חלקי
- ב־Type I המוטציה רצסיבית; ב־DN היא דומיננטית
פתרון
התשובה הנכונה היא (2).
מהשקפים (שקפים 73, 76-77):
קולגן Type I = Triple helix - שלוש שרשראות (2 × α1 + 1 × α2).
OI Type I (Haploinsufficiency - שקף 76): מוטציה שגורמת ל־Null allele ← האלל הפגום לא מייצר שרשרת ← 50% כמות קולגן, אבל הקולגן שנוצר תקין ← OI קל (שברים חוזרים, סקלרות כחולות).
OI חמור (Dominant Negative - שקף 77): מוטציה (לדוגמה החלפת Glycine) ← שרשרת פגומה משתלבת ב־Triple helix ← הורסת את כל ה־Trimer, גם את השרשראות התקינות. מתוך כל הטרימרים, 7/8 יכילו לפחות שרשרת אחת פגומה ← קולגן לא תפקודי ← OI לטאלי או חמור מאוד.
שאלה 19: אכונדרופלזיה - Gain of Function
מהו המנגנון של Achondroplasia ומדוע רוב המקרים הם de novo?
- חסר בקולגן Type II; 50% de novo כי המוטציה נדירה
- Gain of Function ב־FGFR3 (Hot spot mutation); 80% de novo
- מוטציה רצסיבית בגן הורמון גדילה; 100% מורשת
- חסר בקולטן לסידן בעצמות; 60% de novo
פתרון
התשובה הנכונה היא (2).
מהשקפים (שקפים 79, 82):
Achondroplasia: גננות - הפרעה בגדילת עצמות ארוכות.
המנגנון: מוטציית Gain of Function בגן FGFR3 (Fibroblast Growth Factor Receptor 3). ה־Receptor פעיל באופן קבוע (constitutively active) גם ללא ליגנד ← איתות מוגבר ← עיכוב גדילת סחוס.
Hot spot mutation - אותה מוטציה ספציפית חוזרת שוב ושוב באופן עצמאי.
80% de novo - רוב המקרים מתרחשים כמוטציה חדשה בגמטות של הורים בריאים.
Thanatophoric Dysplasia (שקף 86): מוטציה חמורה יותר ב־FGFR3 ← לטאלית.
שאלה 20: אנטיסיפציה ו־Trinucleotide Repeats
מהי אנטיסיפציה (Anticipation) ומה מאפיין אותה?
- מחלה שמופיעה מוקדם יותר בכל דור עקב מוטציה חדשה בכל פעם
- רצפי חזרות לא יציבים שמתארכים בכל דור - מחלה חמורה יותר ומוקדמת יותר
- דילוג דורות - המחלה מופיעה בדור סב ונכד אבל לא באב
- ירידה בחומרת המחלה לאורך הדורות עקב סלקציה טבעית
פתרון
התשובה הנכונה היא (2).
מהשקף (שקף 93):
Anticipation: “Repeats are unstable. Longer with each generation. More severe clinical presentation.”
בפדיגרי שבשקף: מספר החזרות גדל לאורך הדורות - 55 ← 75/85 ← 90/85/85 ← 235/250/260/240/250. ככל שמספר החזרות גדול יותר ← גיל הופעה מוקדם יותר ומחלה חמורה יותר.
דוגמה: Huntington Disease (שקף 94) - חזרות CAG בגן HTT. מעל 36 חזרות ← מחלה. Myotonic Dystrophy, Fragile X - דוגמאות נוספות.
חשוב לייעוץ גנטי: הסיכון לצאצא מושפע לא רק מהגנוטיפ ההורי אלא גם מנטיית ההתארכות של החזרות.