פתיחה - איפה אנחנו נמצאים?
השיעור ממשיך את שני השיעורים הטכניים על ספריות DNA ו־Next Generation Sequencing. בשיעור הקודם דיברנו על החשיבות הקלינית של NGS, על Sanger sequencing, על העקרונות שהופכים את NGS ליעיל, ועל הכנת ספריית DNA: סלקציה למקטעים הרצויים, פרגמנטציה, חיבור אדפטורים, Size selection, אמפליפיקציה וריצוף.
השיעור הנוכחי מתמקד בשני חלקים:
- RNA-seq (RNA Sequencing) - איך מכינים ספרייה שמתחילה מ־RNA ולא מ־DNA.
- NGS data analysis - מה עושים עם הדאטה אחרי שהמכונה מחזירה מיליוני רצפים קצרים.
בסוף השיעור יש גם פתיחה קצרה ל־Third-Generation Sequencing, ובעיקר ל־Nanopore sequencing, כפתרון לחלק מהמגבלות של NGS הקלאסי.
RNA-seq
RNA-seq - שימושים
RNA-seq היא אחת מספריות ה־NGS הנפוצות ביותר. כמעט כל מעבדה שעוסקת בביטוי גנים, דיפרנציאציה, סרטן, תגובה לטיפול או אפיון רקמות יכולה להשתמש בה.
המטרה המרכזית של RNA-seq היא למדוד רמות ביטוי של גנים. כלומר, לשאול: כמה RNA נוצר מכל גן בתא או ברקמה מסוימים.
מתוצאות על רמת הביטוי נגזרים מספר שימושים:
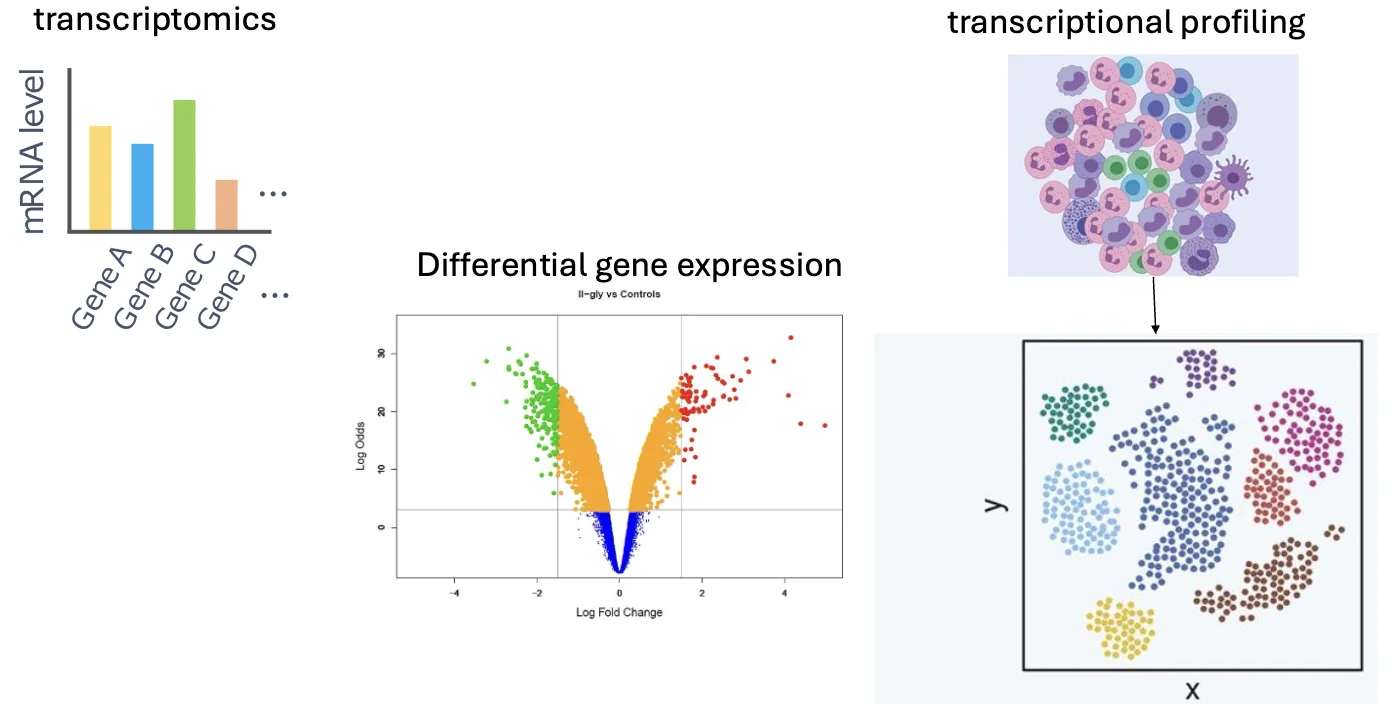
- Transcriptomics analysis - מדידה רחבה של רמות הביטוי של גנים רבים או של כלל הגנים בדגימה.
- Differential gene expression - השוואה בין שני תנאים או יותר, למשל תא סרטני לעומת תא נורמלי, תא מטופל לעומת תא לא מטופל, תא צעיר לעומת תא זקן.
- Single-cell RNA-seq - ריצוף RNA ברמת התא הבודד, בעיקר כאשר הדגימה היא רקמה הטרוגנית שמכילה כמה סוגי תאים.
RNA-seq - Bulk לעומת Single-cell
ב־bulk RNA-seq לוקחים דגימה שלמה ומרצפים את ה־RNA מכל התאים יחד. לכן, מה שמתקבל הוא ממוצע של ביטוי הגנים בכל אוכלוסיית התאים.
ב־single-cell RNA-seq מנסים לקבל פרופיל ביטוי לכל תא בנפרד. זה חשוב במיוחד בדגימות הטרוגניות, כמו דם או רקמה שמכילה הרבה סוגי תאים. כך ניתן לדעת גם אילו סוגי תאים קיימים בדגימה וגם מה כל סוג תא מבטא.
לדוגמה, אם מתקבל פרופיל ביטוי שמתאים ללימפוציטים, אפשר להסיק שיש בדגימה אוכלוסיית לימפוציטים ואף להעריך את החלק היחסי שלה בדגימה. עם זאת, הזיהוי נשען על ידע קודם: צריך לדעת איך נראה פרופיל ביטוי של סוגי תאים שונים.
RNA-seq - השלבים הטכניים
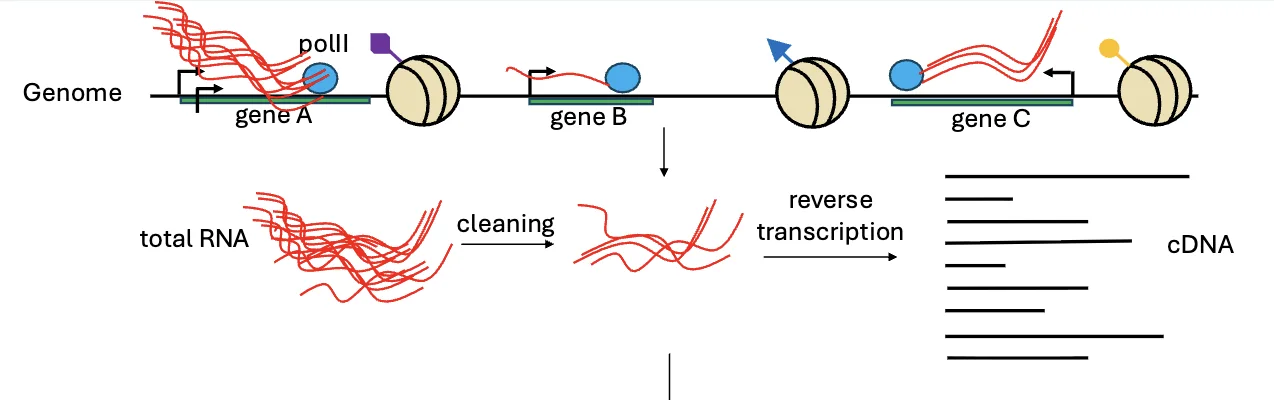
ב־RNA-seq מתחילים מ־RNA, אבל מכונת הריצוף בסופו של דבר מרצפת DNA. לכן צריך להפוך את ה־RNA ל־cDNA, ורק לאחר מכן להמשיך לשלבים הרגילים של הכנת ספריית DNA.
השלבים העיקריים:
- הפקת total RNA מהדגימה.
- ניקוי/סלקציה של RNA - להישאר עם סוגי ה־RNA שמעניינים אותנו.
- Reverse transcription - המרת RNA ל־cDNA.
- Fragmentation - חיתוך המקטעים לפי הצורך.
- Adapter ligation - חיבור אדפטורים.
- Size selection - בחירת מקטעים בטווח גודל מתאים.
- PCR amplification - אם צריך מספיק חומר לריצוף.
- Sequencing - ריצוף מקבילי במכונה.
סוגי RNA וניקוי הדגימה
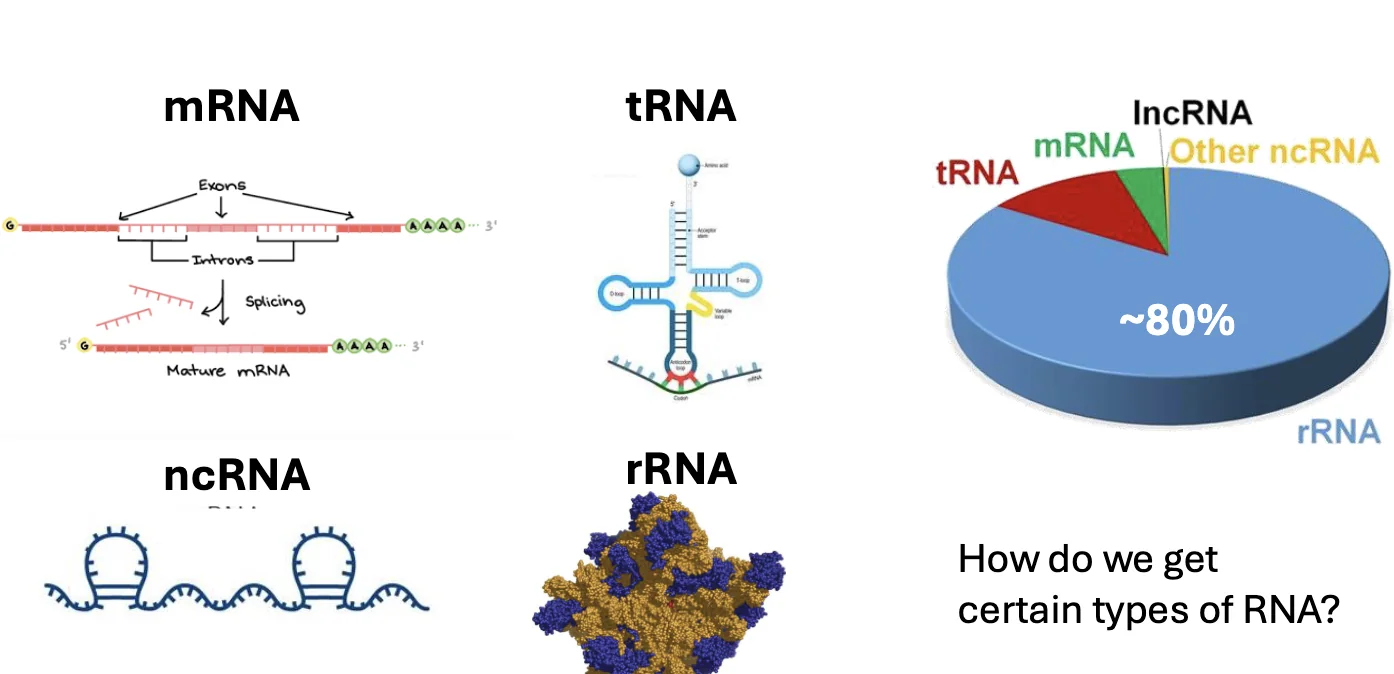
כאשר מפיקים RNA מתא, לא מקבלים רק mRNA. מקבלים תערובת של סוגי RNA שונים.
1. mRNA
mRNA הוא RNA מקודד לחלבון. זה בדרך כלל סוג ה־RNA שמעניין אותנו ב־RNA-seq, כי הוא משקף ביטוי של גנים.
מאפיינים חשובים:
- עובר splicing - אינטרונים מוסרים ואקסונים מחוברים.
- יש לו 5’ cap.
- יש לו poly-A tail בקצה.
2. Non-coding RNA
ncRNA הוא RNA שאינו מקודד לחלבון. בעבר נטו לחשוב עליו כעל פחות חשוב, אבל כיום ברור שיש לו תפקידים רגולטוריים רבים, גם בשעתוק וגם בתרגום.
לכן, לפעמים נרצה לרצף גם ncRNA - תלוי בשאלת המחקר.
3. tRNA ו־rRNA
בדרך כלל, tRNA ו־rRNA אינם מעניינים אותנו ב־RNA-seq רגיל. הבעיה המרכזית היא בעיקר rRNA, משום שהוא מהווה בערך 80% מה־RNA בתא, ולעיתים אף יותר בדגימות מסוימות.
אם לא נסיר rRNA, רוב הקריאות בריצוף יגיעו ממנו. זה בזבוז של עומק ריצוף: במקום לקבל מידע על גנים שמעניינים אותנו, נקבל המון קריאות שמגיעות מ־RNA ריבוזומלי, שהוא בעיקר מבני ולא אינפורמטיבי לשאלת ביטוי הגנים.
חשוב לדייק: זה לא ש־rRNA “לא חשוב” לתא. הוא חשוב מאוד כחלק מהריבוזום. הוא פשוט בדרך כלל לא אינפורמטיבי לשאלות של ביטוי גנים וזהות תאית.
שתי גישות לניקוי RNA
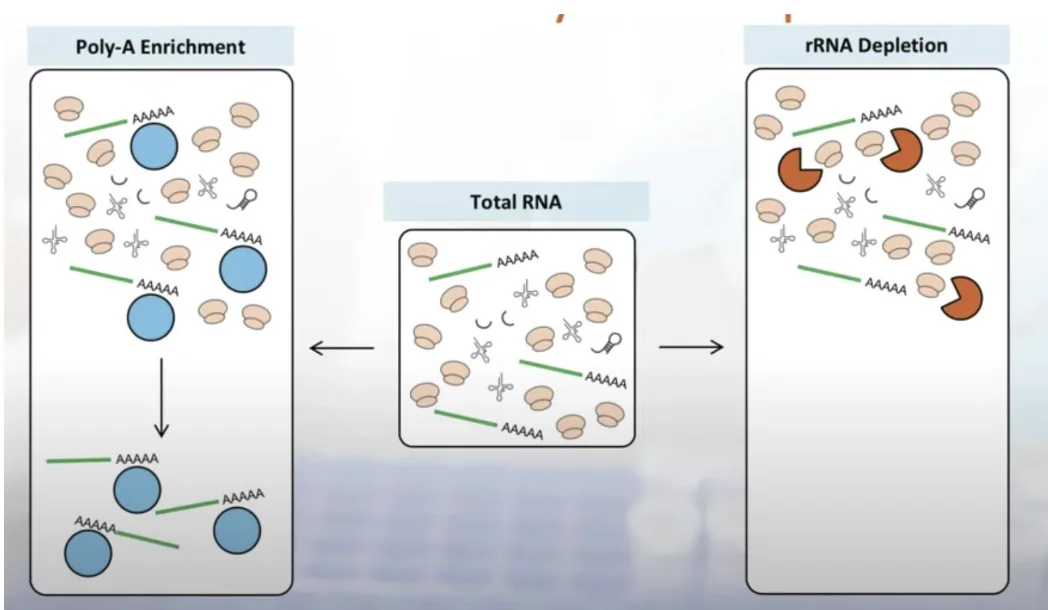
יש שתי גישות עיקריות לסלקציה של RNA לפני RNA-seq:
- rRNA depletion: סלקציה שלילית - מסירים את ה־rRNA ומשאירים את שאר ה־RNA.
- mRNA enrichment / Poly(A) capture: סלקציה חיובית - מבודדים את ה־mRNA לפי ה־poly-A tail.
rRNA Depletion - RiboZero - סלקציה שלילית של RNA ריבוזומלי
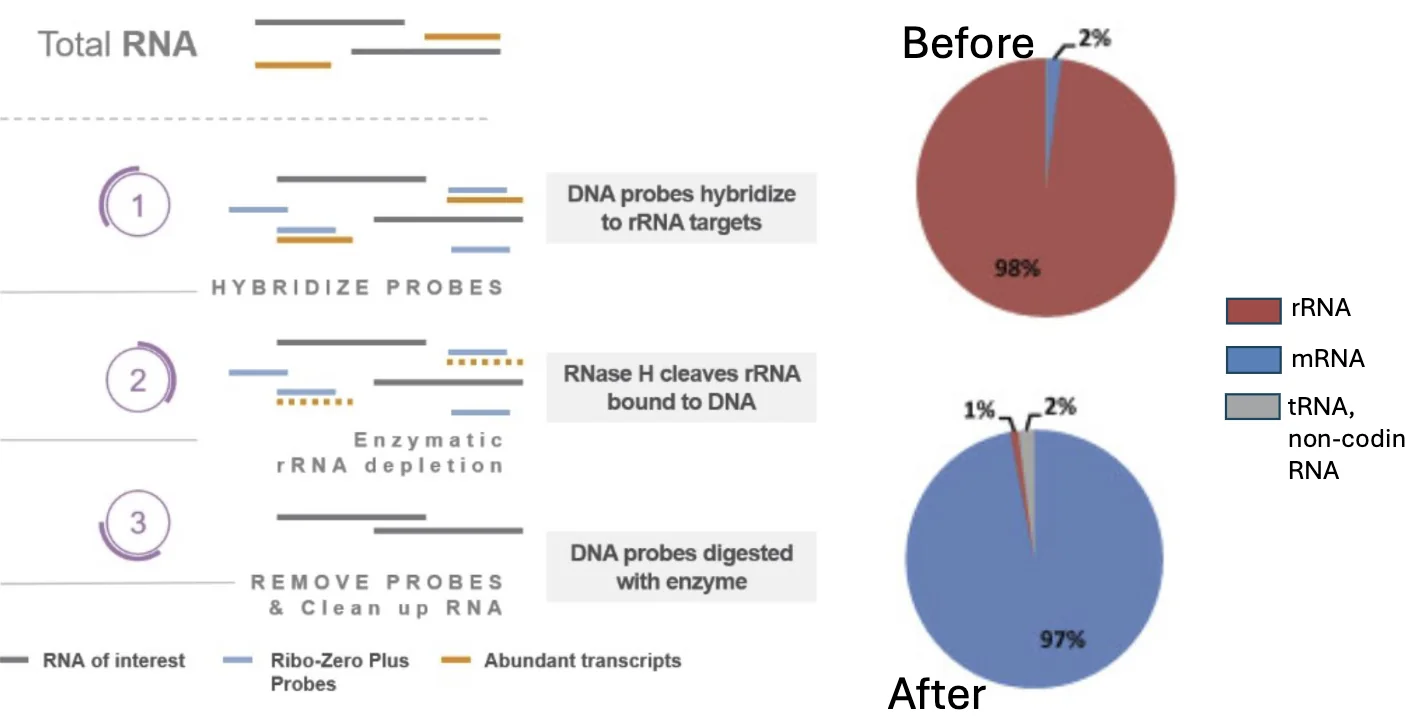
בגישה זו רוצים להיפטר מה־rRNA, אבל להשאיר את שאר סוגי ה־RNA בדגימה.
העיקרון:
- מוסיפים DNA probes שתוכננו כך שיהיו קומפלמנטריים לרצפים ספציפיים ב־rRNA.
- הפרובים נקשרים ל־rRNA ויוצרים DNA-RNA hybrid.
- האנזים RNase H מזהה RNA שנמצא בתוך היבריד DNA-RNA.
- RNase H מפרק את ה־rRNA שקשור לפרובים.
- בסוף נשארת דגימה שהוסר ממנה רוב ה־rRNA.
הגישה הזאת מנצלת תכונה פיזיולוגית של RNase H: הוא יודע לזהות RNA שקשור ל־DNA. במעבדה משתמשים בתכונה הזאת כדי להסיר באופן ממוקד rRNA.
למה זה חשוב?
מספר הקריאות במכונת הריצוף מוגבל. גם אם יש מאות מיליוני reads, עדיין כל read עולה כסף ותופס מקום בדאטה. לכן לא רוצים “לבזבז” את רוב הריצוף על rRNA.
אם הדגימה לא מספיק נקייה, נקבל מעט מאוד מידע על ה־RNA הנדיר יותר, שדווקא עשוי להיות זה שמעניין אותנו.
mRNA Enrichment - Poly(A) capture
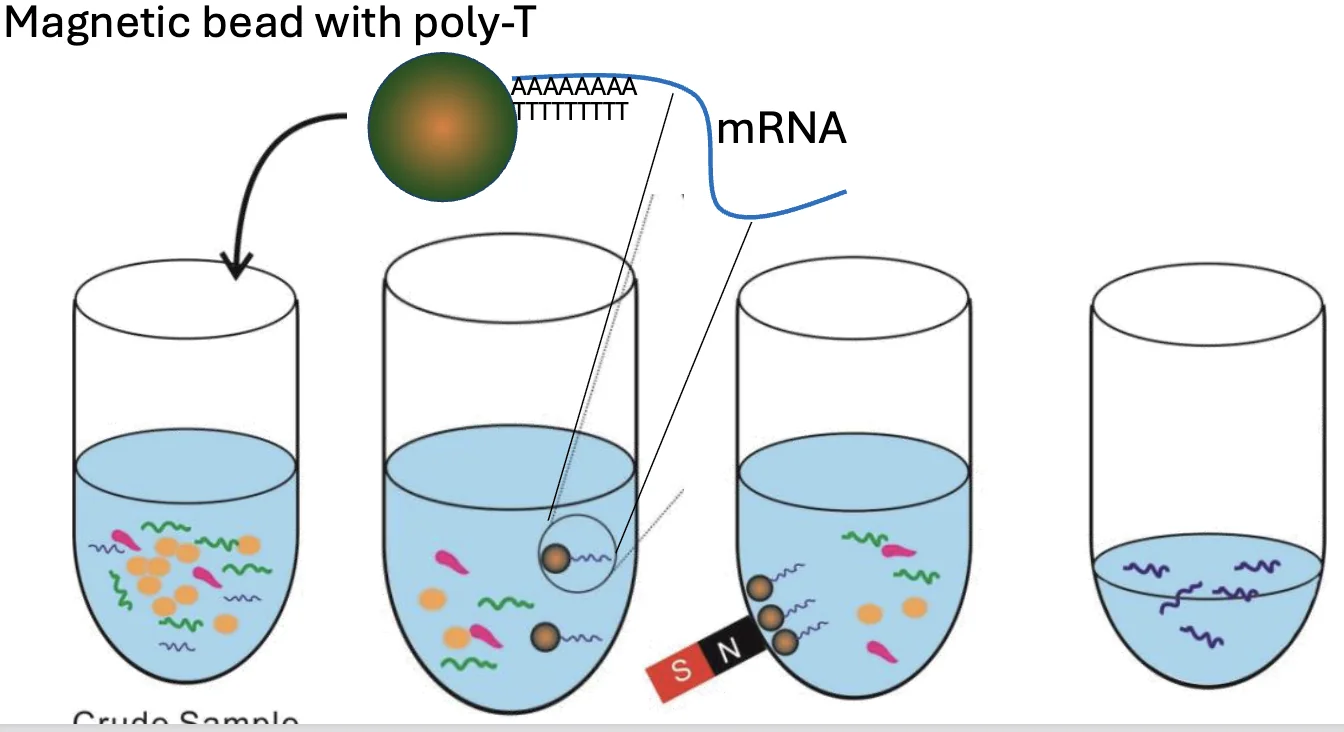
בגישה זו לא מסירים את מה שלא רוצים, אלא דגים את מה שכן רוצים: mRNA.
העיקרון מבוסס שוב על בידים מגנטיים:
- משתמשים בבידים מגנטיים שמצופים ב־poly-T.
- ה־poly-T עובר היברידיזציה עם ה־poly-A tail של mRNA.
- מפעילים מגנט ושולפים את הבידים יחד עם ה־mRNA שנקשר אליהם.
- שוטפים את שאר ה־RNA שלא נקשר.
- נשארים בעיקר עם mRNA.
כמו בשיעור הקודם, השאלה החשובה בבידים מגנטיים היא תמיד: מה התכונה שעליה עושים סלקציה? כאן התכונה היא poly-A tail.
בדיקה עצמית: איזה עוד תכונות ראינו בשיעור הקודם לעניין סלקציית בידים מגנטיים?
פתרון
- Size selection - DNA גדול יותר יותר שלילי ולכן יותר נקשר לקבוצת קרבוקסיל בבידים.
- ChIP-seq - הספציפיות של הבידים היא לחלבון, באמצעות נוגדן.
- mRNA enrichment - הספציפיות של הבידים היא ל־poly-A tail, באמצעות poly-T.
מתי משתמשים בכל גישה?
| גישה | מתי נשתמש בה? | היגיון |
|---|---|---|
| rRNA depletion | כשרוצים לרצף RNA שאינו בהכרח poly-A, למשל ncRNA | מסירים רק את rRNA ומשאירים את שאר סוגי ה־RNA |
| rRNA depletion | בדגימות פרוקריוטיות | ב־RNA פרוקריוטי אין poly-A tail כמו ב־mRNA איקריוטי רגיל, ולכן Poly(A) capture לא יעבוד טוב |
| mRNA enrichment / Poly(A) capture | כשמטרת הניסוי היא transcriptomics רגיל של mRNA | זו סלקציה חיובית ל־mRNA ולכן בדרך כלל נקייה וממוקדת יותר |
| mRNA enrichment / Poly(A) capture | כשחשוב להפחית זיהומים שאינם poly-A | כל מה שאין לו poly-A tail לא יידגם בצורה יעילה |
באופן כללי, כאשר המטרה היא רמות ביטוי של mRNA באאוקריוטים, Poly(A) capture היא ברירת המחדל. כאשר רוצים גם non-coding RNA, או כאשר עובדים עם RNA פרוקריוטי, rRNA depletion מתאים יותר.
הבדל עקרוני: בסלקציה שלילית יודעים מה הסרנו, אבל לא בהכרח יודעים בדיוק מה נשאר. בסלקציה חיובית בוחרים ישירות את מה שרוצים.
ספריית NGS - המסלול הכללי
לאחר ניקוי ה־RNA, מבצעים Reverse transcription ומקבלים cDNA. מכאן ממשיכים כמו בספריות DNA אחרות: חיתוך, חיבור אדפטורים, בחירת גודל, אמפליפיקציה וריצוף.
האדפטורים שמחוברים למקטעים מאפשרים להם להיקשר ל־Flowcell של מכונת הריצוף. אפשר לחשוב על ה־Flowcell כעל משטח שמכיל “שדה” של רצפים קצרים, אליהם האדפטורים יכולים להיקשר. לאחר מכן מתבצע ריצוף מקבילי של כל הספרייה.
NGS data analysis
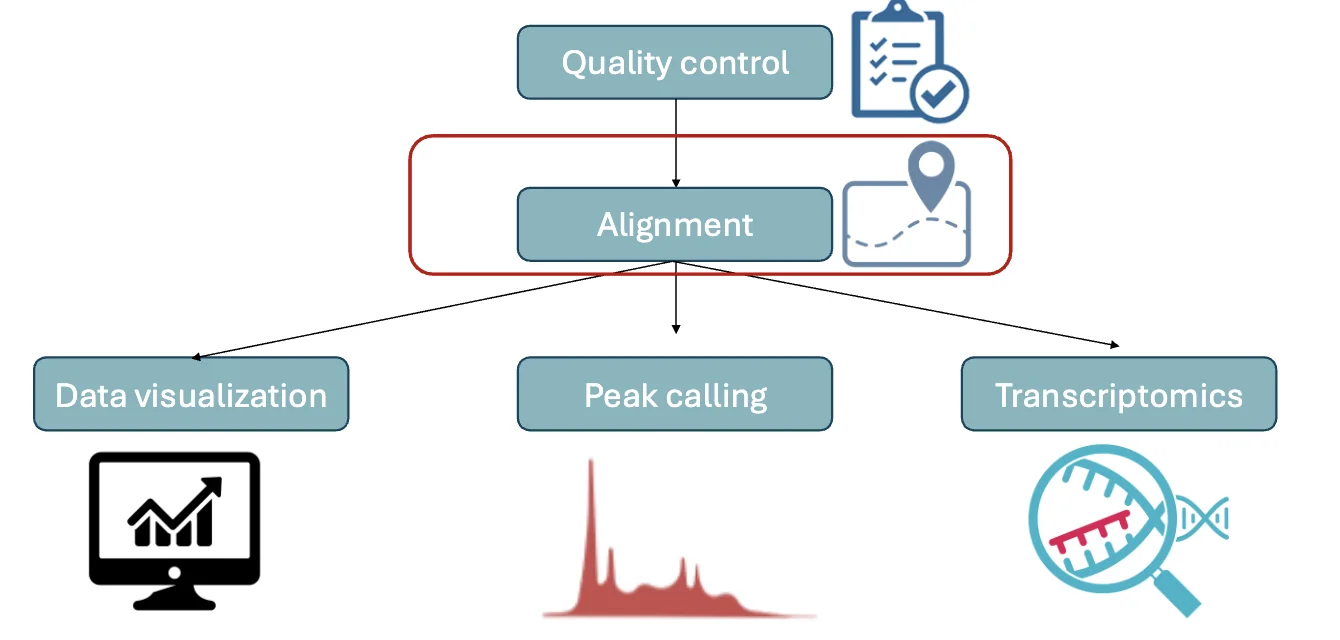
אחרי הריצוף, מתקבלת מהמכונה כמות עצומה של דאטה: רצפים קצרים, לכל אחד איכות קריאה, ולעיתים עשרות גיגה של מידע לכל דגימה.
התהליך הכללי של אנליזת NGS:
- Quality control - לבדוק את איכות הקריאות ולסנן reads באיכות נמוכה.
- Alignment - למפות כל read למיקום המתאים בגנום או בטרנסקריפטום.
- Data visualization - להסתכל על הדאטה בצורה ישירה, למשל ב־genome browser.
-
Downstream analysis - תלוי בסוג הספרייה:
- Peak calling ב־ChIP-seq או ATAC-seq.
- Transcriptomics analysis ב־RNA-seq.
- Variant calling כשמחפשים מוטציות או וריאנטים.
Sequence alignment
שתי דרכים להרכיב פאזל
אפשר לחשוב על alignment כמו על הרכבת פאזל. יש שתי אסטרטגיות שונות:
- להשוות חלקים זה לזה - לחבר חתיכות לפי דמיון בין הקצוות שלהן.
- להשוות כל חלק לתמונה מלאה - לקחת כל חתיכה ולשאול איפה היא מתאימה בתמונה שכבר ידועה.
ב־NGS רגיל, בדרך כלל עובדים לפי השיטה השנייה. יש לנו reference genome, כלומר רצף גנום ידוע, ואנחנו ממפים אליו את ה־reads שקיבלנו מהריצוף.
De-novo genome assembly לעומת alignment לרפרנס
ב־de-novo genome assembly מנסים לבנות גנום מחדש בלי להסתמך על רפרנס. זה דומה להרכבת פאזל בלי תמונה. צריך לחבר רצפים לפי חפיפה ביניהם, ולכן רצפים ארוכים עוזרים מאוד.
לעומת זאת, ב־sequence alignment יש לנו גנום רפרנס. לכן השאלה היא לא “איך לבנות את הגנום?”, אלא “איפה כל read נמצא בגנום הידוע?”.
בגלל ש־Illumina ו־NGS קלאסי מייצרים בדרך כלל short reads, הם מתאימים מאוד למיפוי לרפרנס, אבל פחות מתאימים להרכבת גנום חדש מאפס.
הפלט הראשוני - FASTQ

הקובץ שמתקבל מהריצוף נקרא בדרך כלל FASTQ file. זהו קובץ טקסט גדול שמכיל את הקריאות ואת איכות הקריאה שלהן.
אפשר לחשוב על FASTQ כעל קבוצות של ארבע שורות לכל read:
- שורת כותרת - מזהה של ה־read ומידע טכני על מקורו.
- שורת הרצף - האותיות A, C, G, T, ולעיתים N כאשר המכונה לא הצליחה לזהות בסיס.
- שורת הפרדה
- שורת איכות - קידוד של מידת הביטחון של המכונה בכל בסיס.
הדבר החשוב ביותר לזכור מה־FASTQ הוא שהוא נותן שני סוגי מידע:
- מה הרצף שנקרא.
- מה איכות הקריאה.
לכן לפני alignment מסננים reads באיכות נמוכה. אם read באיכות נמוכה, יש סיכוי גבוה יותר שהוא מכיל שגיאות טכניות.
Reference Genome File

מול קובץ ה־FASTQ עומד reference genome: רצף DNA ידוע של הגנום, למשל אדם או עכבר.
פעולת ה־alignment שואלת עבור כל read:
- באיזה כרומוזום הוא נמצא?
- מה נקודת ההתחלה והסוף שלו?
- באיזה גדיל הוא יושב - הגדיל החיובי או הגדיל המשלים?
הפלט של alignment הוא כבר לא רק רשימת רצפים, אלא רשימה של מיקומים גנומיים.
איך מחפשים בגנום שלם?
האתגר החישובי ברור: איך מחפשים רצף קצר בתוך גנום של שלושה מיליארד בסיסים, ועוד עושים זאת עבור מיליוני reads?
חיפוש ליניארי פשוט היה איטי מדי. לכן משתמשים ב־genome indexing.
האנלוגיה מהשיעור היא אינדקס בסוף ספר: במקום לקרוא את כל הספר בכל פעם שמחפשים מילה, משתמשים באינדקס שאומר באילו עמודים היא מופיעה.
באופן דומה, אפשר להפוך את גנום הייחוס (reference genome) למבנה אינדקס שמאפשר למצוא רצפים בצורה יעילה בהרבה (העיקרון מזכיר hash tables, אבל לא נכנסנו לפרטים חישוביים).
אתגרים ב־Alignment
Multiple alignments
בעיה אחת היא שקריאה (read) יכולה להתאים ליותר ממקום אחד בגנום. זה קורה במיוחד באזורים חוזרניים, אבל יכול לקרות גם בגלל רצפים קצרים מדי.
כל קריאה הגיעה פיזית ממולקולה אחת וממיקום אחד, אבל מבחינה חישובית ייתכן שהיא מתאימה למספר מיקומים. לכן נוצר חוסר ודאות.
הפתרון הראשון הוא reads ארוכים יותר. ככל שה־read ארוך יותר, יש יותר מידע, ולכן קל יותר לדעת מאיפה הוא הגיע. בעבר reads היו קצרים יותר; כיום Illumina יכולה לרצף בדרך כלל עשרות בסיסים עד סביב 100 בסיסים באיכות טובה. מעבר לכך האיכות יורדת.
Paired-end reads
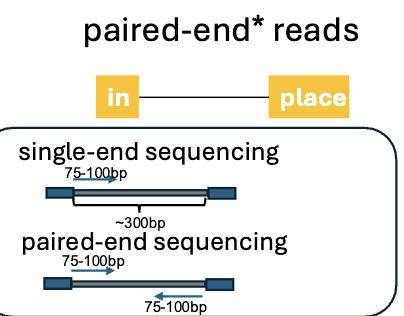
דרך נוספת להקטין אי ודאות היא paired-end sequencing.
בספרייה יש לנו פרגמנטים של DNA, למשל סביב 300 בסיסים. לא חייבים לרצף את כל הפרגמנט. אפשר לרצף 75–100 בסיסים מצד אחד, וגם 75–100 בסיסים מהצד השני.
כך מקבלים שני reads מאותו פרגמנט, ויש גם מידע על המרחק הצפוי ביניהם. אם read אחד מתאים לכמה מקומות, אבל read שני מתאים רק לאחד מהם במרחק הנכון ובאוריינטציה הנכונה, הוודאות עולה.
הרעיון: אם read אחד לא מספיק כדי למפות חד־משמעית, שני קצוות מאותו פרגמנט נותנים יותר הקשר.
Mismatches
גם אחרי סינון איכות, לא תמיד תהיה התאמה מושלמת בין read לבין רפרנס.
יש לכך שתי סיבות עיקריות:
- שגיאת קריאה טכנית - המכונה קראה בסיס לא נכון.
- שוני אמיתי מה־reference genome - פולימורפיזם טבעי, SNP, או מוטציה שנרכשה במהלך החיים.
לכן אלגוריתמים של alignment מאפשרים בדרך כלל מספר קטן של mismatches. למשל, אפשר לאפשר שניים או שלושה mismatches ב־read קצר, ועדיין לקבל alignment סביר.
אבל יש איזון: אם מאפשרים יותר מדי mismatches, עלולים למפות reads למקומות לא נכונים. אם מאפשרים מעט מדי, נאבד reads אמיתיים שיש בהם שונות ביולוגית או שגיאה טכנית קטנה.
RNA-seq alignment
ל־RNA-seq יש אתגר ייחודי שקשור לאינטרונים. הספרייה נבנית מ־cDNA, שמקורו ב־mRNA שעבר splicing. לכן ב־cDNA אין אינטרונים. לעומת זאת, ה־reference genome כן מכיל אינטרונים.
כתוצאה מכך, read יכול להגיע משני אקסונים שהיו רחוקים זה מזה בגנום, אבל התחברו זה לזה ב־mRNA לאחר שחבור (gapped reads). אם ננסה למפות read כזה ישירות לגנום, הוא לא בהכרח יתאים לרצף אחד רציף.
פתרון 1: alignment ל־reference transcriptome
הפתרון הפשוט הוא למפות את reads ל־reference transcriptome ולא לכל הגנום. במקום לשאול איפה ה־read נמצא בגנום, שואלים איפה הוא נמצא ברשימת הטרנסקריפטים הידועים. זה פתרון פשוט ונוח כאשר המטרה היא למדוד רמות ביטוי של גנים מוכרים.
החיסרון: השיטה ״עיוורת״ למה שלא קיים ברפרנס. אם יש transcript לא טיפוסי או Alternative splicing שלא מופיע ברפרנס, ייתכן שלא נזהה אותו.
פתרון 2: split-read / splice-aware alignment
כאשר רוצים לזהות גם אירועי splicing או טרנסקריפטים לא צפויים, אפשר להשתמש באלגוריתמים שמסוגלים להתמודד עם reads מפוצלים.
העיקרון:
- קודם ממפים את reads שניתן למפות בצורה רגילה.
- נשארים עם reads בעייתיים שלא מופו.
- מנסים לפצל אותם ולבדוק אם חלק אחד מתאים לאקסון אחד וחלק אחר מתאים לאקסון אחר.
- משלבים מידע מה־reference genome ומה־reference transcriptome כדי להבין מאיזה splice junction הגיע ה־read.
זו גישה יקרה יותר חישובית, ולכן היא לא תמיד נדרשת. הבחירה תלויה בשאלת המחקר.
אנליזה לאחר Alignment
לאחר alignment, אנחנו יודעים איזה read ממופה לאיזה מקום בגנום או בטרנסקריפטום. בשלב הזה אפשר להתחיל להפיק תובנות ביולוגיות.
האנליזה תלויה בסוג הספרייה:
- ב־ChIP-seq ו־ATAC-seq נחפש אזורי העשרה - כלומר peaks.
- ב־RNA-seq נספור reads לכל גן ונבדוק ביטוי דיפרנציאלי.
- בשיטות אחרות אפשר לחפש וריאנטים, שינויים מבניים או תבניות אחרות.
Data Visualization
לפני סטטיסטיקה, חשוב להסתכל על הדאטה בצורה ישירה. בדרך כלל משתמשים ב־genome browser, למשל UCSC Genome Browser, כדי להעלות את הדאטה ולראות את פיזור הסיגנל לאורך הגנום.
לדוגמה, אם עשינו ChIP-seq למודיפיקציה H3K4me3, שהיא מרקר של פרומוטורים פעילים, נצפה לראות פיקים באזורי התחלת שעתוק. אם הסיגנל מפוזר באופן אקראי או לא נמצא ליד פרומוטורים, ייתכן שהניסוי לא עבד טוב.
בחינה ישירה של הדאטה מאפשרת:
- לבדוק אם הניסוי נראה תקין
- לזהות הבדלים בולטים בין דגימות
- להשוות את הסיגנל לגנים, פרומוטורים, אינהנסרים, SNPs או נתונים מניסויים קודמים
- ליצור היפותזות להמשך בדיקה סטטיסטית
לפעמים עוד לפני האנליזה הכמותית, הדפוס הכללי של הדאטה כבר “קופץ לעין” ומכוון לשאלה הבאה.
Peak calling
מהו Peak calling?
Peak calling הוא ניתוח שמטרתו לזהות אזורים בגנום שבהם יש העשרה של סיגנל.
במילים פשוטות: האלגוריתם עושה את מה שהעין עושה כשמסתכלים על genome browser - מזהה איפה יש פיק - אבל עושה זאת בצורה שיטתית וסטטיסטית.
Peak calling מתאים לספריות שבהן הסיגנל מופיע כאזורים מועשרים בגנום, למשל:
- ChIP-seq - איפה חלבון מסוים או מודיפיקציית היסטון נמצאים לאורך הגנום.
- ATAC-seq - אילו אזורים בכרומטין פתוחים ונגישים.
ב־RNA-seq רגיל בדרך כלל לא עושים peak calling, כי שם השאלה אינה “איפה יש פיק?”, אלא “כמה reads יש לכל גן?”.
הפלט של Peak calling
הפלט יכול להיות למשל BED file שמכיל קואורדינטות גנומיות של הפיקים. כל שורה היא אזור שלם שבו נמצאה העשרה, ולא read בודד.
כלומר, אחרי alignment יש לנו מיליוני reads. אחרי peak calling יש לנו רשימה מצומצמת יותר של אזורים גנומיים שבהם יש סיגנל מובהק.
למה צריך ביקורת ב־ChIP-seq?
ב־ChIP-seq חשוב מאוד להשוות את הדגימה לביקורת (control), כדי להבדיל בין פיק אמיתי לבין רעש טכני או אזור בעייתי בגנום.
ביקורות אפשריות:
-
Input control - לפני האימונופרציפיטציה שומרים חלק מהדגימה בצד ומרצפים אותו בלי סלקציה לנוגדן. זה נותן תמונת רקע של פיזור הקריאות בלי ChIP. אם אזור מסוים מופיע כפיק גם בקלט (input) וגם בדגימת ה־ChIP, ייתכן שזה לא קישור אמיתי של הפקטור, אלא ריצוף יתר או בעיה טכנית באזור גנומי מסוים.
-
Non-specific antibody control - אפשר להשתמש nugdan שאינו מכוון לאפיטופ ספציפי, כדי להעריך רקע שנובע מהנוגדן או מהתהליך עצמו.
-
Knockout control - אם יש אפשרות לעבוד עם תאים שבהם הפקטור אינו קיים, זאת ביקורת חזקה מאוד. משתמשים באותו נוגדן ובאותו תהליך, אבל הפקטור לא אמור להיות בדגימה. לכן פיקים שנשארים גם בביקורת חשודים כרקע.
מה עושים עם פיקים?
לאחר Peak calling אפשר לשאול כמה שאלות ביולוגיות:
- אילו פיקים משותפים בין שני תנאים?
- אילו פיקים קיימים רק בתא סרטני ולא בתא נורמלי?
- האם הפיקים נמצאים בפרומוטורים, באינהנסרים, בגנים או באזורים אחרים?
- האם הפיקים קשורים למסלולים ביולוגיים מסוימים?
- האם במרכז הפיקים יש מוטיב רצפי משותף?
Motif Analysis
אם מדובר בפקטור שעתוק, אפשר לקחת את רצפי ה־DNA במרכז הפיקים ולחפש מוטיב משותף. כך ניתן ללמוד איזו העדפה רצפית יש לפקטור.
ב־sequence logo, גובה האות בכל עמדה משקף את מידת הוודאות או ההעדפה לאותו בסיס. אם בעמדה מסוימת כמעט תמיד מופיע T, האות T תהיה גבוהה. אם יש חלוקה בין C ו־G, שתי האותיות יופיעו בגובה דומה.
ניתוח כזה יכול לעזור לשאול האם מוטציה בפקטור שעתוק משנה את הספציפיות שלו, או האם בתאים סרטניים יש שינוי באזורים שאליהם הוא נקשר.
Transcriptomics ו־Differential Expression
ב־RNA-seq האנליזה המרכזית היא בדרך כלל Transcriptomics. לאחר alignment או מיפוי לטרנסקריפטום, סופרים כמה reads ממופים לכל גן בכל דגימה.
הפלט הבסיסי הוא טבלה:
- שורות = גנים
- עמודות = דגימות
- בכל תא = מספר reads שמופה לגן הזה בדגימה הזאת
לאחר מכן ניתן לבצע Differential Expression Analysis: השוואה בין תנאים כדי לזהות גנים שעלו או ירדו בביטוי.
Volcano plot
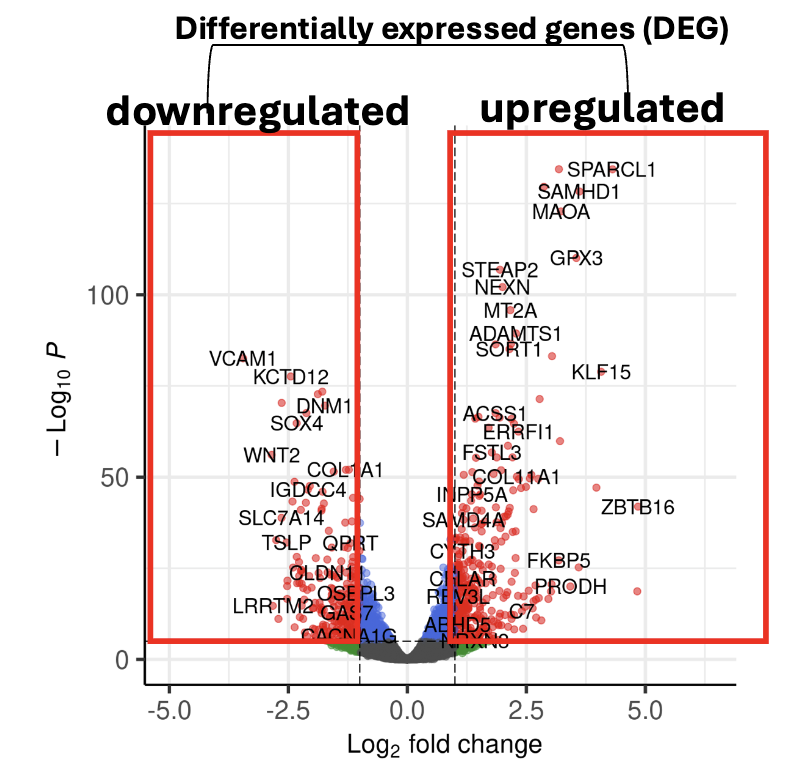
בדרך כלל מציגים Differential Expression ב־Volcano plot.
הציר האופקי הוא לרוב log2 fold change:
- ערך חיובי: הגן מבוטא יותר בתאי הניסוי
- ערך שלילי: הגן מבוטא פחות בתאי הניסוי
הציר האנכי קשור למובהקות סטטיסטית, בדרך כלל p-value מותאם.
כך אפשר לזהות Differentially Expressed Genes (DEGs):
- Up-regulated genes - גנים שעלו בביטוי
- Down-regulated genes - גנים שירדו בביטוי
לאחר שמקבלים רשימת DEGs אפשר לבצע אנליזות המשך, למשל Pathway analysis: לבדוק אם הגנים שעלו או ירדו שייכים למסלול מסוים, כמו דלקת, פרוליפרציה או אפופטוזיס.
Third-Generation Sequencing - Nanopore
בסוף השיעור דיברנו בקצרה על Third-Generation Sequencing, ובעיקר על Nanopore sequencing.
השאלה היא למה צריך דור שלישי אם NGS כל כך יעיל. התשובה היא של־NGS הקלאסי יש כמה מגבלות. בעיקר:
- PCR bias - לא כל המקטעים עוברים אמפליפיקציה באותה יעילות.
- Short reads - רצפים קצרים בעייתיים באזורים חוזרניים ובקישור בין וריאנטים על אותה מולקולה.
מגבלה 1 - PCR bias
בחלק מהספריות יש שלב של PCR. זה בעייתי כאשר רוצים אנליזה כמותית מדויקת, כי PCR יכול להכניס bias.
הבעיה היא שלא כל המקטעים עוברים אמפליפיקציה באותה יעילות. למשל, GC content משפיע על יעילות PCR:
- אזורים עשירים מדי ב־GC יכולים להיות קשים לדנטורציה.
- אזורים עם תכונות רצף אחרות יכולים לעבור אמפליפיקציה בצורה פחות יעילה.
ככל שעושים יותר מחזורי PCR, כך ה־bias גדל. לכן, גם בשיעור הקודם הודגש שכדאי להשתמש בכמה שפחות מחזורי PCR - רק המינימום שנדרש לקבלת מספיק חומר לריצוף.
מגבלה 2 - short reads
היתרון של NGS הוא ריצוף המוני של reads קצרים. אבל זה גם חיסרון.
Short reads בעייתיים במיוחד בשני מצבים:
- אזורים חוזרניים - read קצר לא תמיד מאפשר לדעת מאיזה עותק של הרצף החוזר הוא הגיע.
- קישור בין וריאנטים על אותה מולקולה - אם שני אללים או שני וריאנטים נמצאים באותה מולקולת DNA, short reads עלולים לקרוא כל אחד בנפרד, בלי לדעת שהם נמצאים יחד.
Long reads יכולים לפתור חלק מהבעיות האלה, כי הם מאפשרים לקרוא מקטע ארוך יותר ברצף אחד.
Nanopore sequencing - העיקרון
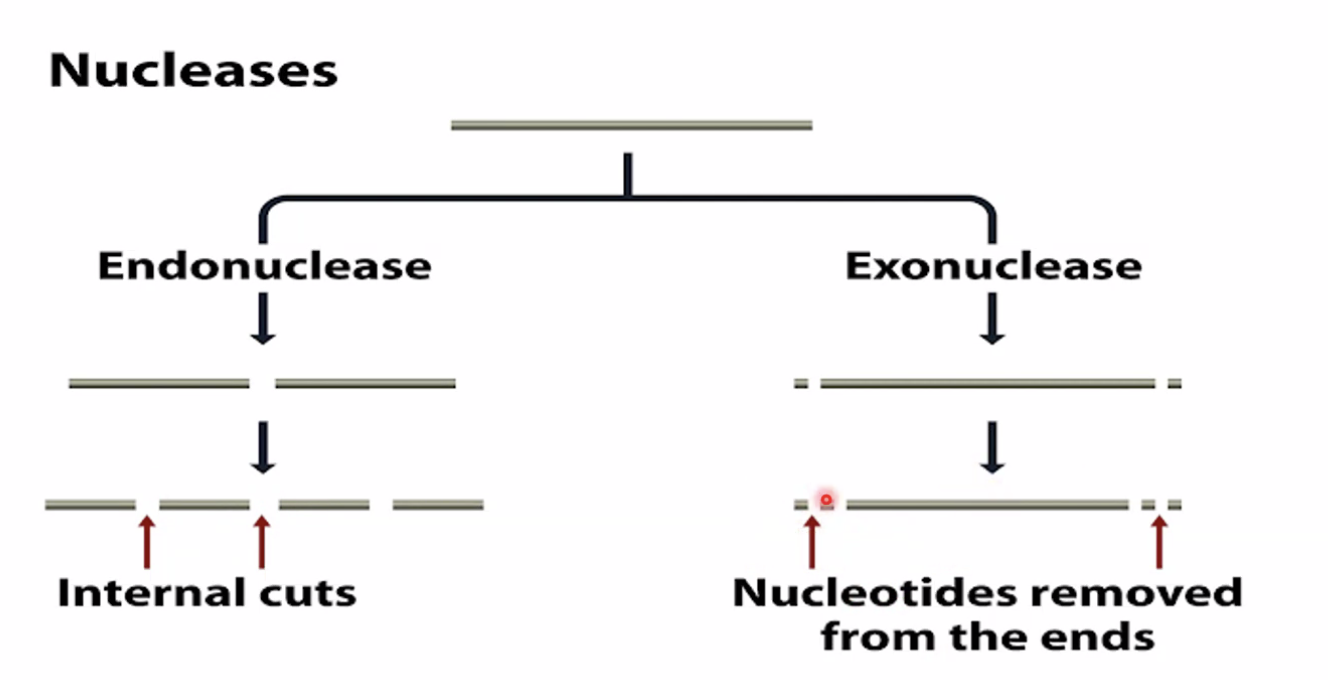
ב־Nanopore sequencing משתמשים בתעלות זעירות שדרכן עוברת מולקולת DNA חד־גדילית. כאשר נוקלאוטיד עובר דרך התעלה, הוא משנה את זרם היונים שעובר בה.
העיקרון:
- DNA מוכנס למערכת עם nanopores.
- הליקאז או רכיב דומה פותח את ה־DNA ומעביר גדיל דרך התעלה.
- כל נוקלאוטיד מפריע לזרם היוני בצורה מעט שונה.
- המכשיר מודד את השינוי בזרם.
- לפי תבנית השינויים החשמליים מסיקים את רצף הבסיסים.
כלומר, בניגוד ל־Illumina, לא קוראים כאן סיגנל פלואורסצנטי בזמן סינתזה, אלא קוראים סיגנל חשמלי בזמן מעבר המולקולה דרך תעלה.
יתרונות
- ניתן לקבל reads ארוכים.
- המכשור יכול להיות קטן ונייד יחסית.
- ניתן לעבוד עם כמויות DNA נמוכות יחסית.
- מתאים במיוחד לשאלות שבהן חשוב לעבור דרך אזורים חוזרניים או להבין קשר בין וריאנטים על אותה מולקולה.
חסרון מרכזי
הדיוק נמוך יותר ביחס ל־Illumina. לכן Nanopore מתאים במיוחד כאשר השאלה אינה דורשת את הדיוק הגבוה ביותר בכל בסיס, אלא מידע על מבנה ארוך יותר: אזורים חוזרניים, וריאנטים רחוקים על אותה מולקולה, או ארגון של מקטעים ארוכים.
סיכום
RNA-seq היא ספריית NGS שמתחילה מ־RNA, אבל חייבת לעבור המרה ל־cDNA כדי להיכנס לעולם הריצוף. השלב הקריטי לפני ההמרה הוא סלקציה לסוג ה־RNA שמעניין אותנו: או שמעשירים mRNA בעזרת Poly(A) capture, או שמסירים rRNA בעזרת rRNA depletion.
אחרי הריצוף, האתגר עובר מהמעבדה למחשב. ה־FASTQ נותן רצפים ואיכות קריאה, אבל כדי להפיק מהם משמעות צריך לבצע quality control, alignment, להסתכל בדאטה ולערוך אנליזות המשך.
- ב־ChIP-seq וב־ATAC-seq: מחפשים פיקים - אזורים מועשרים בגנום.
- ב־RNA-seq: סופרים reads לפי גנים ומחפשים ביטוי דיפרנציאלי.
לכל ספרייה יש downstream analysis שמתאים לשאלה הביולוגית שלה.
לבסוף, Third Generation Sequencing, ובמיוחד Nanopore, נועד להתמודד עם חלק מהמגבלות של ה־NGS הקלאסי: PCR bias ו־short reads. היתרון הגדול שלו הוא reads ארוכים, גם אם הדיוק הבסיסי נמוך יותר.
דור פסקלשורת תחתונה: הכנת הספרייה קובעת איזה מידע נכנס לריצוף. ה־alignment קובע איפה המידע שרוצף נמצא בגנום. האנליזה הביולוגית קובעת מה המשמעות של הדאטה.