1) איך התחילו להבין שווירוס יכול לגרום לסרטן (Rous sarcoma virus)
פתחנו בתופעה שנראתה “מוזרה”: בניסוי בתרנגולות הופיע סרטן באופן שנראה מדבק.
פייטון ראוס לקח רקמה סרטנית, טחן אותה, וסינן את התמצית בפילטר שלא מעביר תאים וחיידקים (אבל כן מעביר וירוסים).
את הנוזל המסונן הוא הזריק לתרנגולות, והן פיתחו סרטן. אחר כך הוא חזר על התהליך שוב ושוב (סרטן ← טחינה ← סינון ← הזרקה) וכל פעם התקבל סרטן. זאת הייתה אחת הפעמים הראשונות שהראו שסרטן יכול “לעבור” בצורה שמרמזת על גורם זיהומי.
2) איך מודדים כמה וירוס מדבק יש במבחנה: Plaque assay
כדי לכמת “כמה וירוס מדבק יש”, משתמשים במבחן הפלאקים:
- מגדלים תאים ב־monolayer, מוסיפים דילולים שונים של הווירוס, ואז מוסיפים שכבת חומר (כמו אגר) שמגבילה את הדיפוזיה כדי שהווירוס לא “ינדוד” חופשי.
- כל חלקיק ויראלי מדבק יתחיל מוקד הדבקה מקומי: הוא ידביק תא, יהרוג אותו, ויתפשט רק לשכנים-עד שרואים חור/אזור מת במונולייר (Plaque).
- ספירת הפלאקים מאפשרת לחשב את כמות הווירוס המדבק.
בווירוס ראוס (Rous sarcoma virus, RSV) יש משהו חריג: הוא לא בהכרח הורג תאים, אבל כן גורם לשינוי מורפולוגי ואף יכול לקדם תהליך סרטני בתרבית.
3) “מהפכת” הרוורס-טרנסקריפטאז: איך הגיעו לזה שלמרות שהגנום RNA, יש שלב DNA
הפרדוקס: בווירוס ראוס הפיקו גנום RNA, אבל ניסויים הראו תלות בשלבי סינתזת DNA.
ניסוי 1: מעכב סינתזת DNA עוצר יצירת וירוס
תיארו ניסוי שבו הוסיפו מעכב של DNA synthesis בשלב ההדבקה־ואז בהמשך לא נוצר וירוס, למרות שמדובר בווירוס RNA. זה רמז שחייב להיות שלב שבו נוצרת מולקולת DNA.
ניסוי 2: היברידיזציה של RNA ויראלי עם DNA של תא מודבק
סימנו את ה־RNA הוויראלי רדיואקטיבית, ועשו היברידיזציה מול DNA מתאי יעד:
- בתא מודבק התקבלה היברידיזציה (יש רצף משלים ב־DNA).
- בתא לא מודבק לא התקבלה היברידיזציה.
המסקנה: לווירוס יש מעבר שבו הוא “משתלב” איכשהו עם DNA תאי.
ניסוי 3: לא צריך סינתזת חלבון כדי להתחיל את התהליך
פוסט-דוקטור הוסיף Cycloheximide (מעכב סינתזת חלבון), ובכל זאת הווירוס הצליח לבצע את תהליך ההיפוך/ההשתלבות. זה הוביל להצעה שהאנזים הדרוש מגיע כבר בתוך הוויריון (ארוז בקפסיד).
שתי מעבדות במקביל + מבחני אנזים במבחנה
במקביל, הווארד טמין ודיוויד בלטימור הגיעו לאותה תגלית: במערכת in vitro ראו תוצר רדיואקטיבי שמתקבל רק אם יש:
- מגנזיום
- דאוקסי-נוקלאוטידים (dNTPs), ולא מספיק רק rNTPs
וכן, כשהרסו את התוצר עם DNase (אבל לא עם RNase) זה הראה שהתוצר הוא DNA.
מכאן נולד המושג: Reverse transcriptase - אנזים שמייצר DNA על תבנית RNA, ומשנה את הדוגמה הקלאסית.
אפשר לקשר את זה ישירות לטכנולוגיות מודרניות: היום אפשר לקחת RNA (למשל מדם), להפוך אותו ל־cDNA ואז לעשות PCR/RT-PCR וריצוף RNA-וכל זה “יושב” על התגלית הזו.
4) מחזור חיים בסיסי של רטרו־וירוסים: כניסה, יצירת dsDNA, אינטגרציה, יציאה
המהלך הכללי:
- הווירוס נכנס לתא (פיוז’ן לממברנה), נכנס קפסיד.
- Reverse transcriptase מייצר DNA דו־גדילי.
- ה־DNA נכנס לגרעין ומשתלב בגנום התא בעזרת Integrase.
- מרגע שהווירוס משולב (provirus), הוא יכול להישאר כחלק מהגנום ולהתבטא בעת הצורך.
- התא משתמש במנגנוני התא (RNA polymerase II, capping, splicing, polyA) לייצור RNA ויראלי וחלבונים.
- אריזה על הממברנה, יציאה כוויריון חדש.
Simple vs Complex retroviruses
ההבדל:
- Simple retroviruses: מתקשים להיכנס לגרעין בצורה פעילה ← אינטגרציה בעיקר בתאים מתחלקים (כשמעטפת הגרעין “נפתחת” בחלוקה).
- Complex retroviruses (למשל לנטי-וירוסים): יכולים להכניס את ה־DNA גם לתאים לא מתחלקים. זו הסיבה שבמחקר, כשרוצים להכניס גנים לנוירונים משתמשים בלנטי-וירוסים.
5) נקודות שחייבים לזכור על רטרו־וירוסים
- הגנום הוא RNA חיובי.
- הוא דיפלואידי: יש שני עותקים של RNA באותו ויריון (סוג של “גיבוי” אם אחד נשבר).
- הגנום לא משמש ישירות כתבנית לסינתזת DNA תאי; הוא קודם עובר הפיכה ל־DNA דו־גדילי.
- תא שנדבק יכול לשאת את גנום הווירוס לנצח בגלל האינטגרציה.
כמה מהגנום שלנו הוא “עקבות” של רטרו־אלמנטים?
בערך 45% מהגנום שלנו קשור לרטרו־אלמנטים/טרנספוזונים שנכנסו לאורך האבולוציה. דוגמאות:
- בצמחים זה יכול להגיע לכמויות קיצוניות (גם 95% בהקשר מסוים).
- צוין אלמנט בשם LINE-1 שמוערך בכ־17% מהגנום, ומתבטא בעובר; עיכוב הביטוי שלו פוגע בהתפתחות עוברית.
6) שלוש הפעילויות של Reverse transcriptase
שלוש פעילויות אנזימטיות עיקריות:
- RNA-dependent DNA polymerase: יצירת גדיל DNA על תבנית RNA.
- RNase H activity: פירוק ה־RNA מתוך ההיבריד RNA-DNA כדי להשאיר DNA מתאים להמשך.
- DNA-dependent DNA polymerase: סינתזת הגדיל השני לקבלת dsDNA.
פריימר: שימוש ב־tRNA תאי
האנזים צריך primer, וברטרו־וירוסים משתמשים ב־tRNA מהתא כפריימר. לכל רטרו־וירוס יש התאמה לרצף tRNA מסוים (לדוגמה: HIV משתמש תמיד באותו סוג tRNA).
בנוסף לכך, “kissing loop” - אזור קומפלימנטרי שמחזיק שני עותקי ה־RNA יחד.
7) “הקפיצות” במנגנון הרוורס-טרנסקריפשן ולמה מכפילים קצוות (LTR)
המנגנון:
- מתחילים מסינתזה על קצה אחד, תוך כדי RNase H מפרק את ה־RNA.
- בגלל שיש רצפים חוזרים בקצוות, ה־DNA שנוצר יכול “לקפוץ” ולהיצמד לקצה השני ולהמשיך סינתזה.
- יש אזור RNA שנשמר (PPT) ומשמש כפריימר לגדיל השני.
- בסוף מתקבל DNA שבו קצוות מסוימים מוכפלים ויוצרים LTR בשני הצדדים.
למה זה קריטי:
ה־RNA הוויראלי שנוצר בתא לא מכיל את כל הרצף הרגולטורי בצורה “כפולה”, ולכן רק דרך מנגנון הקפיצות בהיפוך ל־DNA מתקבל שוב LTR מלא בשני הקצוות-מה שנחוץ לביטוי הווירוס במחזור הבא אחרי אינטגרציה.
8) ביטוי גנים ברטרו־וירוסים: אותו פרומוטר, אותו polyA, הרבה תוצרים
העיקרון:
- כל התעתיקים מתחילים מאזור ה־LTR ומסתיימים באותו polyA,
- אבל מתקבל מגוון תעתיקים ע”י שחבור (Splicing) ו”טריקים” תרגומיים.
דוגמאות לטריקים
- Frameshift בריבוזום: מאפשר לייצר מאותו RNA גם Gag וגם Gag-Pol. זה לא יעיל (קורה בכ־10% מהתרגומים), וזה טוב כי צריך הרבה Gag (מבנה) ומעט אנזימים.
- Alternative initiation: קודון התחלה פחות יעיל גורם לכך שרק חלק קטן מהתרגומים מתחיל שם - ככה מווסתים כמויות.
- גם ב־mRNA תאי יש מבנים שניוניים וחלבוני קישור - RNA אף פעם לא “ערום”.
איך יוצא מהגרעין RNA שלא עבר Splicing?
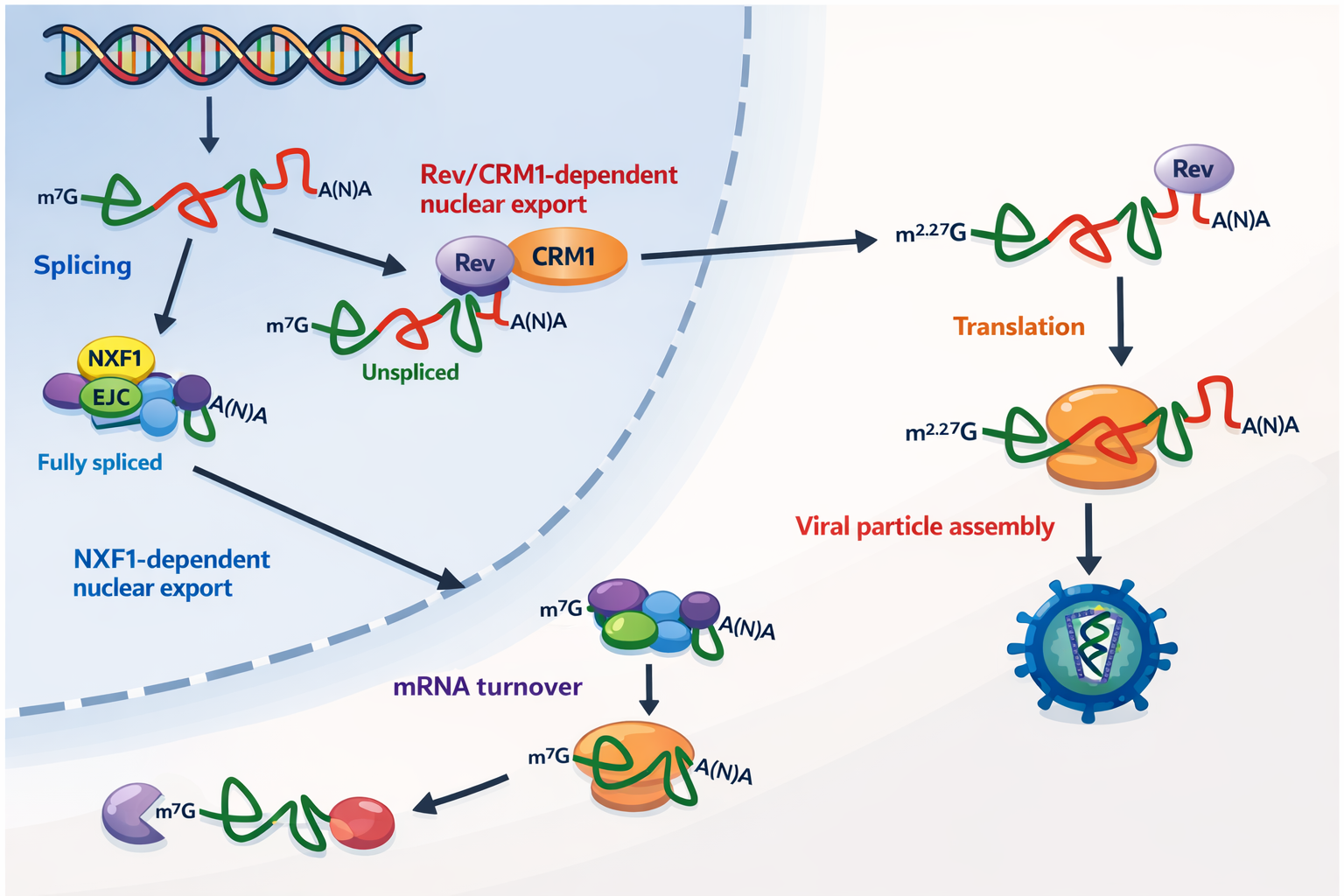
יש בעיה: אם תמיד עושים שחבור, לא יהיה RNA גנומי מלא לאריזה. הפתרון שתואר (במיוחד ב־HIV):
- חלבון Rev נקשר לרצף/לופ ב־RNA, מעכב שחבור,
- ומאפשר ייצוא מהגרעין של RNA לא־מספלייסד דרך חלבון שַׁטְּל (Crm) דרך הנקב הגרעיני.
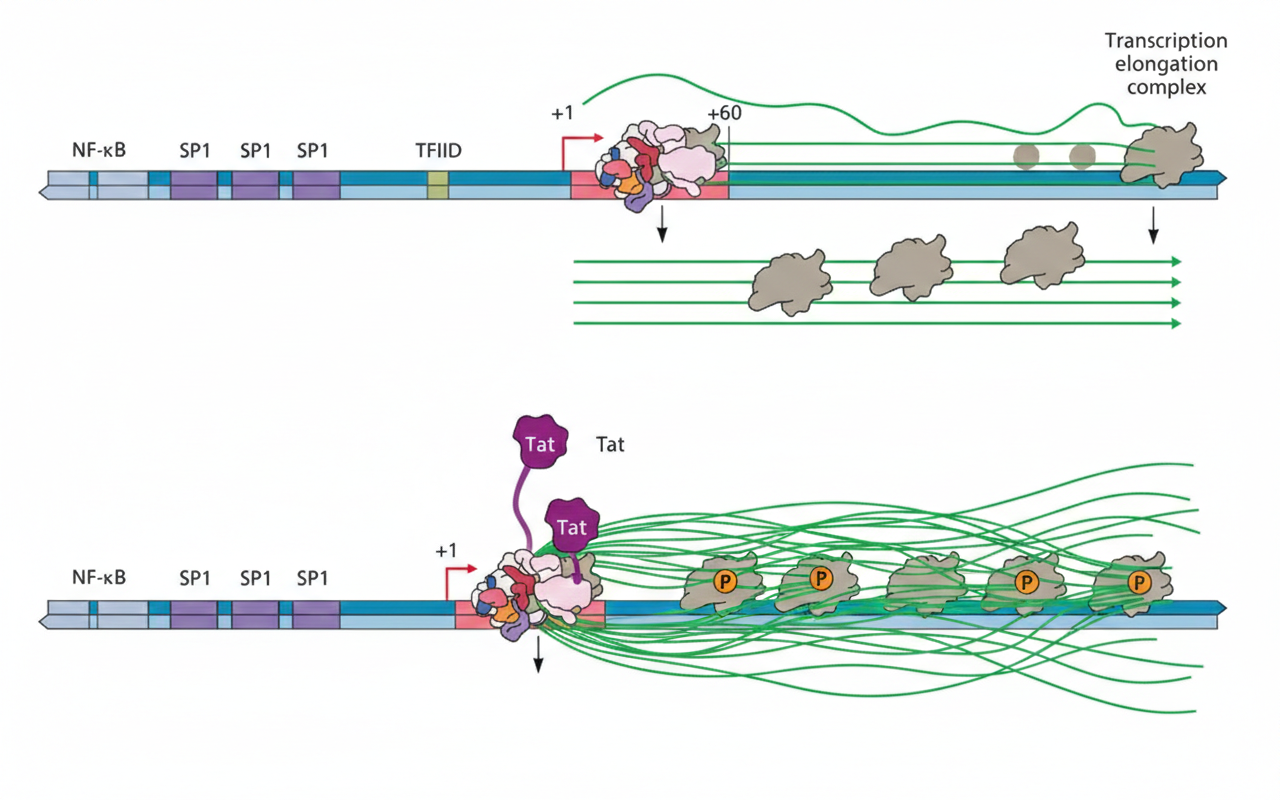
Tat ובקרה על אלונגציה
elongation - הארכת שרשרת תעתוק
Tat הוא חלבון שמקודד על ידי HIV. הוא משמש כרגולטור שמאפשר לפולימראז להמשיך אלונגציה. Tat מגביר את האלוגנציה של הרנ״א הנגיפי:
- בלי
Tatהפולימראז מתחיל תעתוק, יוצר כ־50 נוקלאוטידים, נעצר אחרי capping ונשאר “תקוע”. Tatמגייס קומפלקס שמבצע פוספורילציה ומסיר עיכובים כדי לאפשר elongation.
אפקט
Tatתלוי גם בפקטורים תאיים ספציפיים (למשל, יהיה הבדל בין תאי אדם לתאי עכבר, ובמיוחד יש צורך ב־CyclinT אנושי כדי לקבל תגובה חזקה).
9) אונקוגנים: איך וירוסים “גונבים” גן תאִי וגורמים לסרטן
חזרו ל־Rous sarcoma virus:
- לווירוס יש גן בשם src.
- כשעשו היברידיזציה מצאו הומולוגיה גם בגנום תאים לא מודבקים ← כלומר הווירוס גנב גן מהתא.
- Src הוא חלבון שמעורב בבקרה על חלוקת תאים (פוספורילציה).
- בווירוס הוא הגיע בלי הרגולציה / עם מוטציות שמפעילות אותו קבוע ← תאים מתחלקים ללא עצירה ← טרנספורמציה סרטנית.
מושגים:
- אם המקור ויראלי: v-onc
- אם המקור תאי: c-onc
10) HIV/AIDS: מקור, נתונים, הדבקה, מהלך מחלה, אבחון וטיפול
מקור כללי
בקופים יש וירוסים דומים (SIV), כנראה שחשיפה לדם קופים באפריקה אפשרה מעבר לבני אדם. הוזכר שיש HIV-1 (שכיח יותר) ו־HIV-2 (פחות).
נתונים (בהקשר של 2020)
נתונים מספריים:
- 37 מיליון~ נשאים בעולם.
- 1.5 מיליון~ הדבקות חדשות בשנה.
- 680,000~ נפטרו מאיידס וסיבוכיו.
ריכוז התחלואה והתמותה גבוה באפריקה, גם בגלל זמינות תרופות.
דרכי העברה
הווירוס לא יציב מחוץ לגוף (“כמה שעות”), ולכן ההעברה בעיקר דרך נוזלי גוף:
- מגע מיני
- דם / מוצרי דם (בעבר, לפני בדיקות סקר)
- מחטים מזוהמות (סמים, קעקועים/פירסינג לא סטריליים)
- העברה אנכית אם-לילד (הוזכר)
מהלך המחלה
השלבים:
- הדבקה ראשונית עם וירמיה גבוהה.
- הופעת נוגדנים רק אחרי כמה שבועות (כ־3 שבועות ללא נוגדנים).
- מעבר לפאזה שקטה יחסית (שנים; סדר גודל 7~ שנים, וגם טווח 10-5 שנים בין הדבקה לאיידס).
- בסוף ירידה מתמשכת ב־תאי CD4 T, ובשלב מסוים מופיעים זיהומים אופורטוניסטיים ותסמיני כשל חיסוני.
הוגדר:
- אדם “נורמלי” סביב 500-1200/1500 CD4
- איידס מוגדר כאשר יש פחות מ־200 תאי CD4 במדידת דם.
למה מתים באיידס?
כי מערכת החיסון לא מצליחה לדכא פתוגנים “רגילים”:
- זיהומים חיידקיים
- פטריות
- פרוטוזואה/טפילים
- וגם וירוסים שבדרך כלל נשלטים (כמו CMV והרפס) יכולים להתפשט לאיברים.
יש גם קשר לסרטנים מסוימים כאשר הבקרה החיסונית נשברת.
אבחון ומעקב
- בדיקות נוגדנים: חיוביות רק שבועות אחרי ההדבקה.
- RT-PCR: מפיקים RNA מהדם, הופכים ל־cDNA בעזרת רוורס-טרנסקריפטאז, ומבצעים PCR.
- Real-time RT-PCR משמש למעקב כמותי אחר “כמה וירוס יש בדם” כדי לבדוק יעילות טיפול.
- אפשר לרצף את הווירוס כדי לזהות מוטציות ועמידות לתרופות.
למה אין חיסון
הרוורס-טרנסקריפטאז עושה הרבה טעויות, והווירוס “נהנה” מזה: האנטיגנים משתנים כל הזמן, ויש שונות בין חולים וגם בתוך אותו חולה (“quasispecies”).
קבוצות תרופות/עיכובים
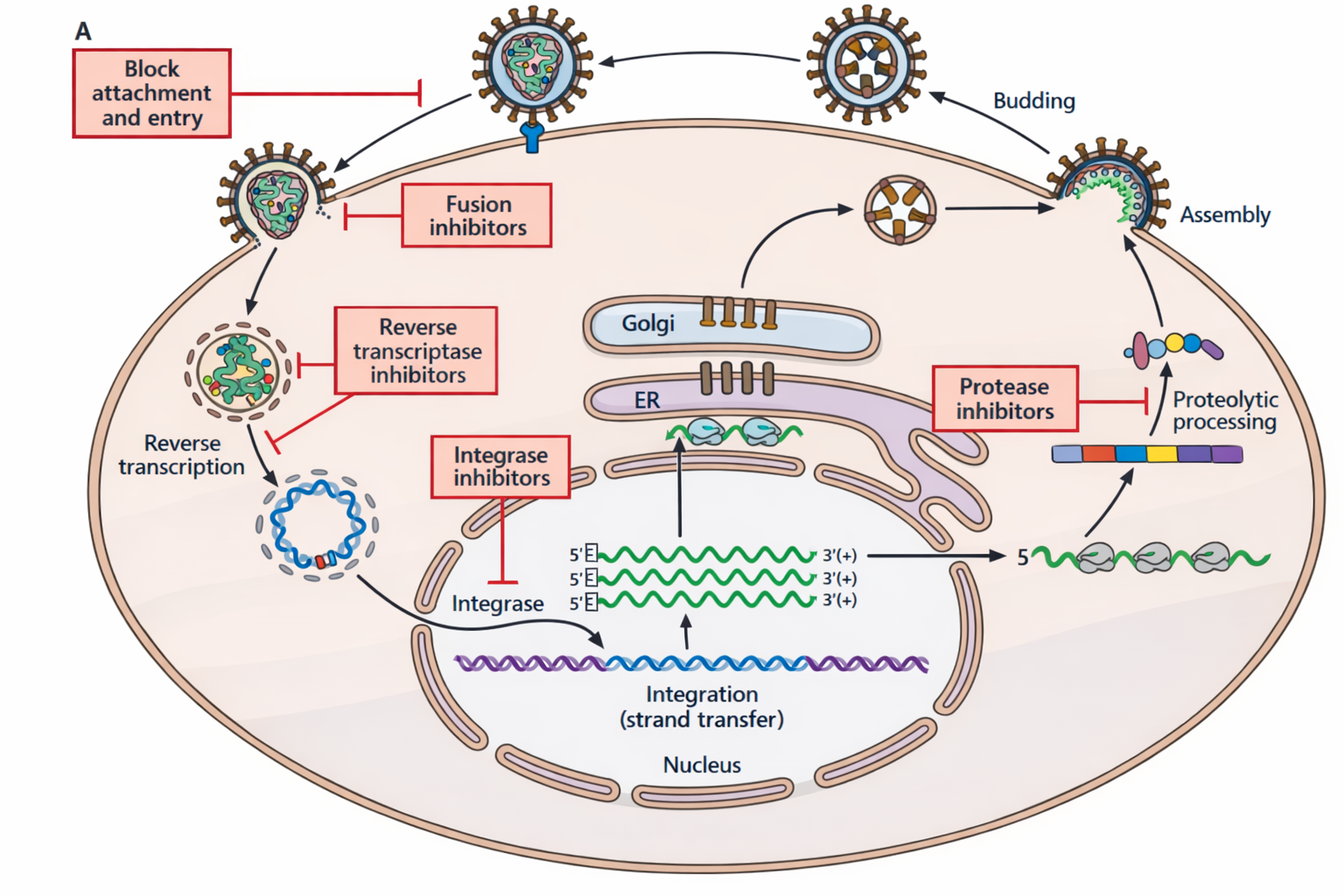
משפחות עיקריות:
- אנלוגים של נוקלאוזידים/נוקלאוטידים: RT “פרומיסקואי” ומכניס נוקלאוטידים לא טבעיים ← עיכוב סינתזה.
- מעכבי RT (עיכוב אתר פעיל).
- מעכבי פרוטאז: בלי ביקוע פוליפרוטאין לא נוצרים חלבונים/אנזימים פעילים.
- מעכבי כניסה/פיוז’ן.
- מעכבי אינטגראז.
- הוזכרו גם כיוונים כמו נוגדנים מנטרלים/עיכוב הבשלה.
עיקרון הטיפול:
לא נותנים תרופה יחידה אלא קוקטייל (שלוש תרופות/מנגנונים שונים), כי הסיכוי לווירוס עם שלוש מוטציות מתאימות באותו גנום נמוך יותר. עוקבים בדם, ואם העומס הוויראלי עולה עושים ריצוף ומתאימים טיפול.
PrEP / PEP והיבט חברתי
הוזכר שיש:
- Post-exposure prophylaxis
- Pre-exposure prophylaxis
יש דיון/מחלוקת חברתית סביב מתן טיפול מונע לאנשים לא חולים מחשש להשפעה על התנהגות סיכון, לצד מצבים שבהם זה כן מועיל (למשל בני זוג בסיכון).
רצפטורים ומקרה “החולה מברלין”
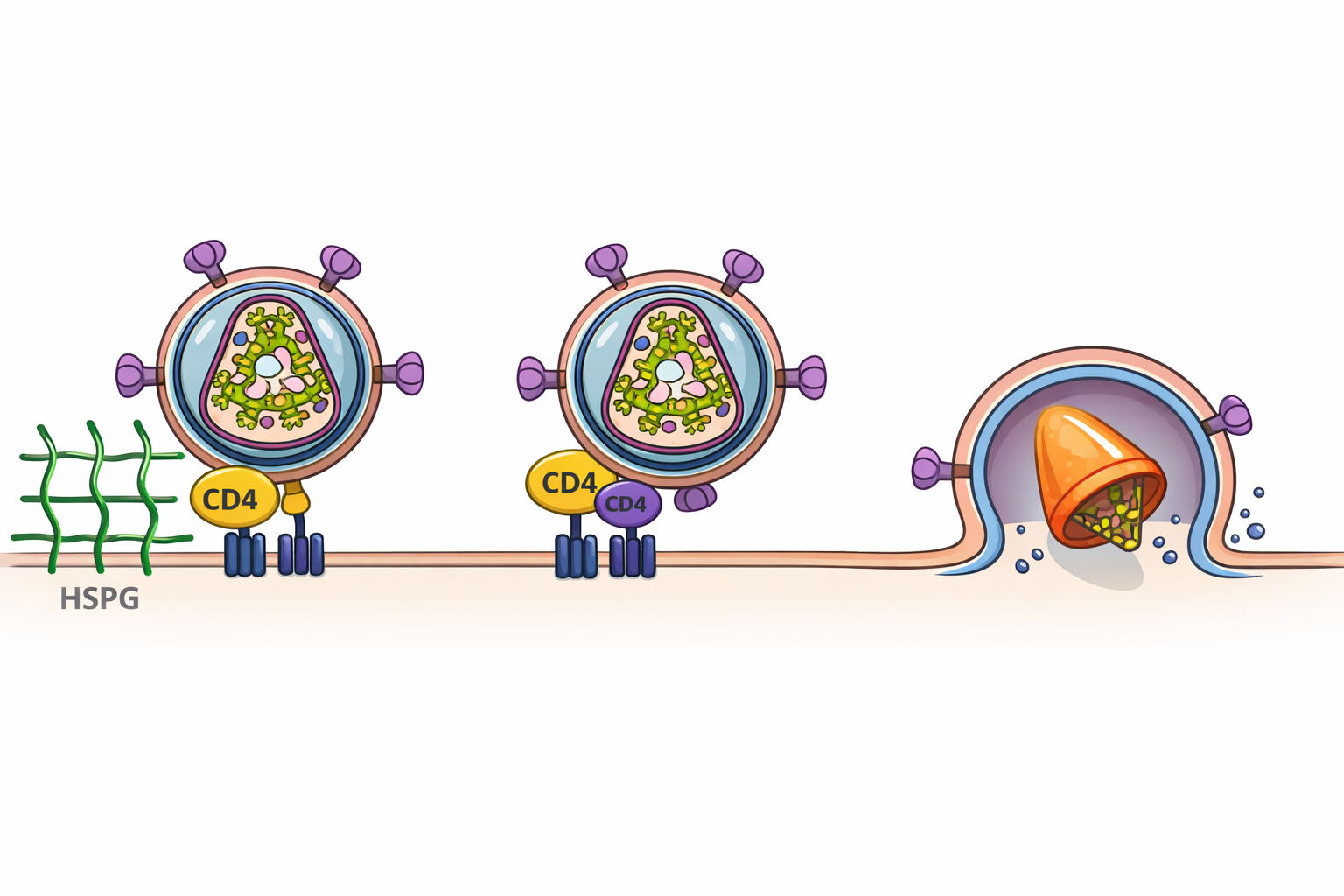
HIV נקשר ל־CD4 וצריך גם קורצפטור CCR5.
יש אחוז קטן באוכלוסייה עם מוטציה ב־CCR5 שלא מאפשרת הדבקה ביעילות.
“החולה מברלין” (Timothy Ray Brown): חולה HIV שפיתח סרטן דם, עבר הקרנה והשתלת מח עצם מתורם עם מוטציית CCR5; לאחר ההשתלה הוא יצא ללא מחלה פעילה, והרעיון הזה השפיע על כיווני מחקר.
כיוונים מחקריים לריפוי
- עריכת גנום בתאי T של החולה כדי לנטרל CCR5 ולהחזיר תאים עמידים.
- גישה של “להעיר” את הווירוס הרדום (latency) ואז לחסל תאים מודבקים (“להוציא אותו מהשקט ואז להרוג”).
המרצה הדגישה: אין “ריפוי” מלא סטנדרטי, אבל עם תרופות אנשים יכולים לחיות שנים רבות בלי קיצור תוחלת חיים, כל עוד נוטלים טיפול.
עיקרון: Undetectable = Untransmittable (U=U) - עומס ויראלי לא מדיד בדם מקטין מאוד הדבקה.
11) HBV (Hepatitis B virus): וירוס DNA קטן עם שלב RT ותלות במנגנוני תא
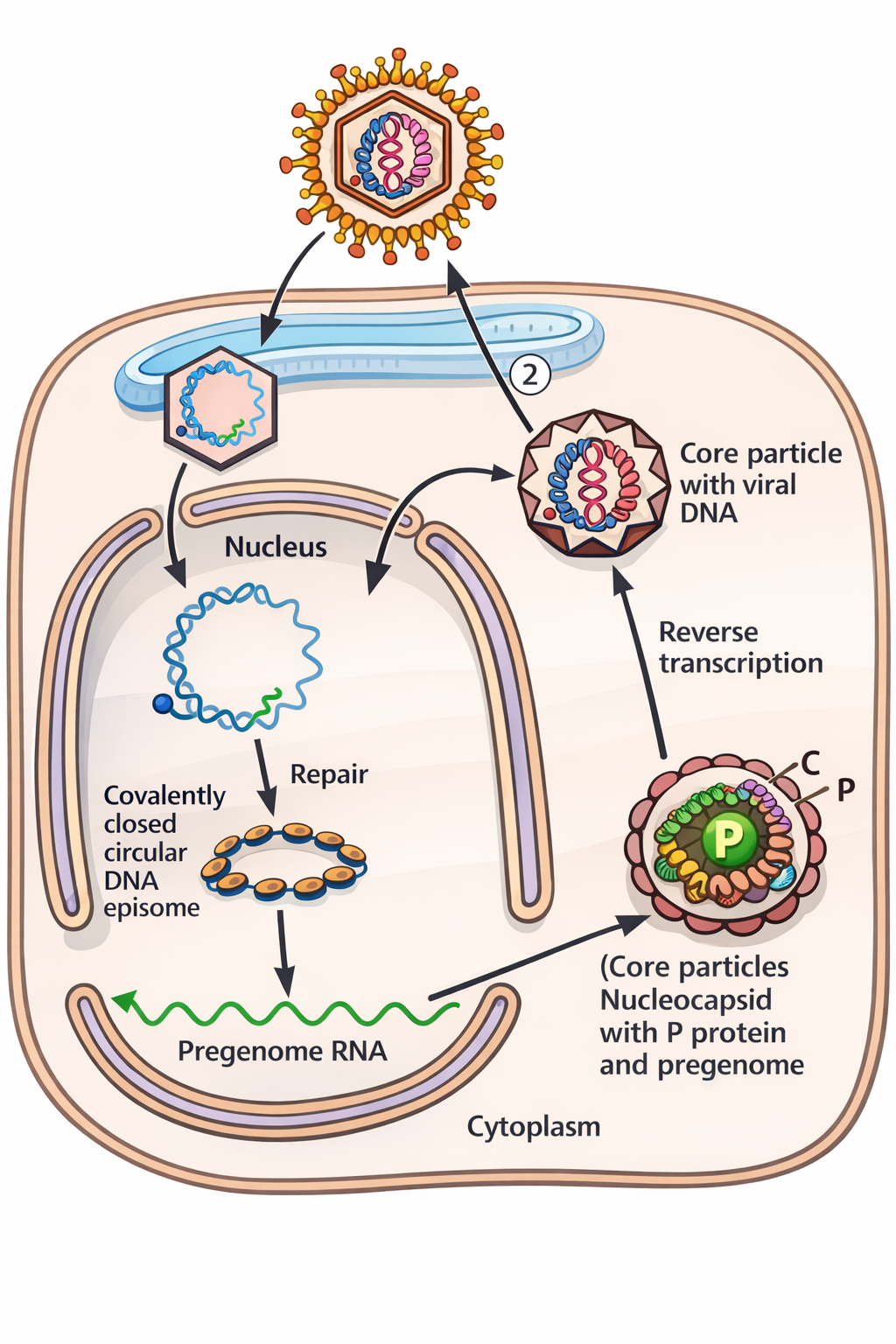
בחלק האחרון תואר HBV:
- הגנום בוויריון הוא DNA מעגלית חלקית דו־גדילית (גדיל אחד מלא, השני חסר).
- לאחר כניסה לתא, מערכת תיקון DNA תאית משלימה אותו ל־DNA דו־גדילי מעגלי בשם cccDNA (אפיזום עטוף נוקלאוזומים).
- מ־cccDNA מתבצע תעתוק ע”י המערכת התאית, ונוצר גם pregenomic RNA.
- בתוך הקפסיד הוויראלי, הפולימראז משתמש ב־pregenomic RNA כדי לייצר שוב DNA (כלומר יש כאן שלב RT בתוך הקפסיד).
- תואר מנגנון שבו הקפסיד “ננעל” אחרי התחלת סינתזת DNA ולכן לא נכנסים עוד נוקלאוטידים ← לכן מתקבל שוב DNA חלקי.
- אם יש מספיק חלבוני מעטפת (surface), הוויריון נעטף ויוצא; אחרת הקפסיד יכול לחזור לגרעין ולהגדיל את מאגר ה־cccDNA (בערך עד 10~ אפיזומים בגרעין תא מודבק).
HBV לא הורג תא “ישירות” - הנזק הוא אימוני
הפגיעה בכבד נובעת בעיקר מתגובה חיסונית נגד הפטוציטים מודבקים: זה גורם לדלקת כבד ולהפרעה בתפקוד (צהבת).
ברוב האנשים מערכת החיסון מחסלת את התאים המודבקים והווירוס נעלם, אבל באחוז מסוים (בעיקר בילדים) זה נשאר כרוני. הוזכר סדר גודל 10-15% כרוני.
איך כרוניות מובילה לצירוזיס ולסרטן
תואר מעגל:
הרג הפטוציטים ← רגנרציה + יצירת רקמת חיבור ← שוב פגיעה ← עוד רקמת חיבור. בסוף מתקבלת צירוזיס וכבד לא פונקציונלי עד צורך בהשתלה.
הוזכר גם סיכון ל־HCC (סרטן כבד) בגלל דלקת כרונית, ובמקרים מסוימים גם אינטגרציה “תקלה” של HBV לגנום (לא חלק הכרחי במחזור החיים) יכולה לתרום לסרטן.
למה HBV ספציפי לכבד
יש ב־HBV enhancers שתלויים בפקטורי תעתוק שקיימים בכבד (דוגמאות: HNF3, HNF4). בתאים שאינם כבדיים אין הפעלה מספקת של הפרומוטורים ולכן אין רפליקציה.
קומפקטיות גנומית קיצונית
גודל הגנום הןא: 3.2kb~ בלבד. בגלל הצפיפות:
- יש שימוש בפרומוטורים שונים, כולם מסתיימים באותו polyA.
- קיימות מסגרות קריאה חופפות: אותו רצף DNA מייצר שני חלבונים שונים כאשר קוראים אותו בהיסט (frame) אחר-מה שממחיש כמה מוטציה אחת יכולה לפגוע בשני חלבונים בו־זמנית.
המרצה גם הזכירה מהיכרות אישית/מחקרית: פותח חיסון מודרני שמבוסס על יצירת מעטפת/חלקיקים ללא גנום, ולא רק “חלבון מעטפת יחיד”.
12) משפטי סיכום
- תגלית הרוורס-טרנסקריפטאז שינתה את כל הביולוגיה המולקולרית והאבחון המודרני של RNA.
- רטרו־וירוסים יכולים להיות לטנטיים שנים, ורק חלק מהתאים נכנסים לפרודוקטיביות.
- HIV קשה לחיסון בגלל קצב מוטציות ושינוי אנטיגני.
- HBV הוא וירוס קטן ודחוס מאוד, והנזק בכבד קשור בעיקר לתגובה החיסונית ולכרוניות.