חזרה על השיעור הקודם
השיעור נפתח בחזרה קצרה על נזקי DNA ברמת הבסיס ועל BER.
הנקודות החשובות:
-
מוטציות
C->Tצפויות במיוחד באזורים שבהם ציטוזין עבר מתילציה.אם 5-methylcytosine (ציטוזין ממוטל) עובר דה־אמינציה, התוצר הוא תימין שהוא בסיס תקין ב־DNA, ולכן קשה יותר לזהות שמדובר בטעות.
-
איבוד בסיס קורה בעיקר בפורינים, אבל גם ציטוזין (C), שהוא פירימידין, יכול להופיע כאתר חסר בסיס כשלב ביניים בתיקון דה־אמינציה.
-
אוקסידציה קשורה באופן ישיר לנשימה תאית ולמיטוכונדריה, דרך יצירת ROS.
-
בתאי B צפויה רמה גבוהה יותר של דה־אמינציה, בגלל האנזים AID שמשתתף בתהליך יצירת וריאציה בנוגדנים.
-
AID יוצר דה־אמינציה, ובהמשך מגויסים מנגנונים פחות מדויקים, ולכן מתקבלת רמת מוטציות גבוהה יותר.
-
הגורם המרכזי ליצירת AP sites הוא הידרוליזה של הקשר הגליקוזידי (N-glycosidic bonds).
-
בתיקון 8-oxoG האנזים הראשון שפועל הוא OGG1.
-
ההבדל בין BER של דה־אמינציה לבין BER של אוקסידציה הוא בגליקוזילאז הראשון: UNG בדה־אמינציה, ו־OGG1 באוקסידציה.
-
במיפוי אתרי 8-oxoG באוקסידציה, משתמשים ב־OGG1 לפני שלב הבידים, כדי להפוך את אתרי האוקסידציה ל־AP sites שאפשר לבודד.
מכאן עברנו מנזקים קטנים ברמת הבסיס ל־bulky lesions.
Bulky DNA Lesions
Bulky DNA lesions הם נזקי DNA שנוצרים בדרך כלל מחיבור קוולנטי של מולקולה ריאקטיבית גדולה לאחד מבסיסי ה־DNA. ההבדל העיקרי בינם לבין ה־non-bulky lesions הוא בגודל ובהשפעה המבנית:
- non-bulky lesions - הנזק קטן יחסית, בדרך כלל ברמת בסיס יחיד, ושלד הסוכר-פוספט נשאר שלם.
- bulky lesions - הנזק גדול מספיק כדי לשנות את מבנה ההליקס של ה־DNA.
השינוי במבנה ההליקס מפריע לתהליכים שדורשים קריאת DNA מסודרת, בעיקר:
- Replication
- Transcription
דוגמאות ל־bulky lesions:
- Pyrimidine dimers, בעיקר T-dimers (thymine dimers), בעקבות חשיפה ל־UV.
- מולקולות גדולות וריאקטיביות כמו platinum agents, למשל cisplatin.
- מולקולות מעישון, כמו benzopyrene, שיכולות להיקשר קוולנטית ל־DNA.
Pyrimidine Dimers ו־UV
Pyrimidine Dimer
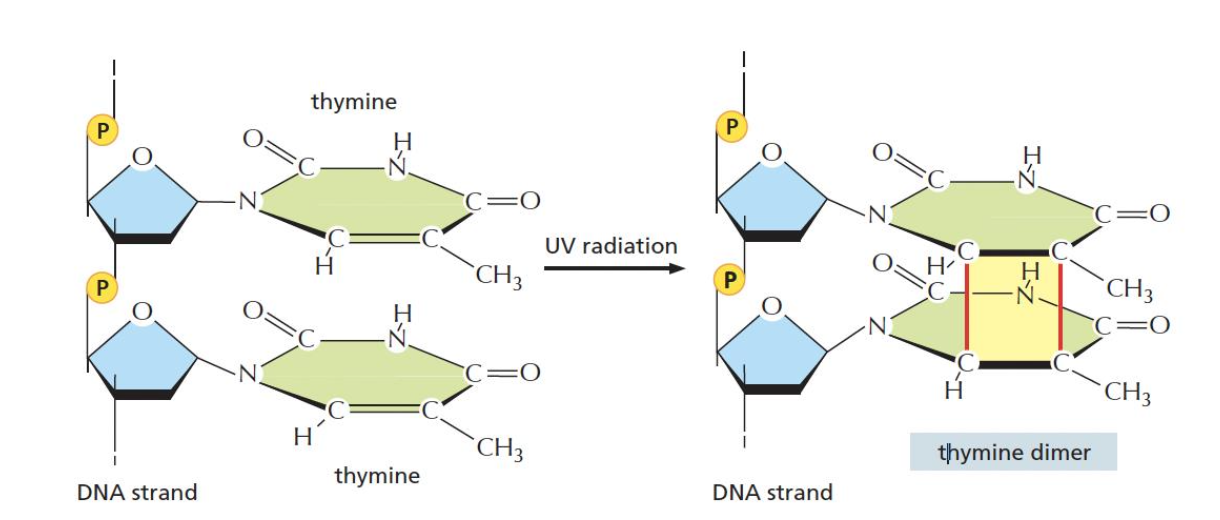
Pyrimidine dimer הוא חיבור קוולנטי לא תקין בין שני פירימידינים סמוכים. הדוגמה הקלאסית היא חיבור בין שני בסיסי תימין סמוכים (T-dimer).
למשל, Cyclobutane Pyrimidine Dimer (CPD).
הנזק הזה נוצר בעיקר בעקבות קרינת UV. ככל שהחשיפה ל־UV חזקה יותר או ממושכת יותר, נוצרים יותר T-dimers.
T-dimer מפריע לשכפול DNA, משום שהוא משנה את המבנה המקומי של ההליקס ומפריע לפולימראז להתקדם.
מיפוי T-dimers בגנום
האם כל אתרי ה־TT בגנום רגישים באותה מידה ליצירת T-dimers? כדי לבדוק זאת צריך למפות אתרים שבהם באמת נוצרו T-dimers, ולא רק לדעת איפה יש רצף TT בגנום.
עיקרון השיטה:
- מקרינים תאים ב־UV כדי לאפשר יצירת T-dimers.
- חותכים את ה־DNA באופן אקראי, למשל בסוניקציה.
- מחברים אדפטורים למקטעים. באדפטור יש ddNTP בקצה כדי לחסום המשך חיבור לא רצוי.
- משתמשים ב־T4 endonuclease וב־APE1 כדי לחתוך באזור ה־T-dimer ולחשוף $\ce{OH}$ חופשי.
- מחברים אדפטור נוסף לאזור שבו נחשף ה־$\ce{OH}$.
- מבצעים PCR וריצוף.
- תחילת הקריאה מסמנת את מיקום הנזק.
כך אפשר להגיע לרזולוציה גבוהה מאוד של מיקום הנזק עצמו.
העשרה באתרי קישור של ETS transcription factors
התוצאה לא טריוויאלית: יש העשרה של T-dimers באתרי קישור של פקטורי שעתוק ממשפחת ETS.
לא מדובר בכך שהרצף עצמו לבדו רגיש יותר. כשעושים את הניסוי על naked DNA (DNA ללא חלבונים שקשורים אליו), ההעשרה לא מופיעה.
ההסבר הוא שהקישור של פקטור השעתוק ל־DNA משנה מעט את המבנה המרחבי של ה־DNA. הוא מקרב את שני בסיסי התימין זה לזה (מקפל), וכך מגביר את הסיכוי שהם יתחברו בקשר קוולנטי לאחר חשיפה ל־UV.
במילים אחרות: חלבוני ETS לא יוצרים את הנזק בעצמם, אלא מסדרים את ה־DNA כך שהנזק ייווצר בקלות רבה יותר כאשר יש UV.
Photoreactivation ופתרונות שאינם קיימים באדם
במהלך האבולוציה הופיעו כמה פתרונות לתיקון T-dimers, בינוניתם:
- Photoreactivation בעזרת האנזים photolyase.
- T4 glycosylase / endonuclease בבקטריופאג’ים.
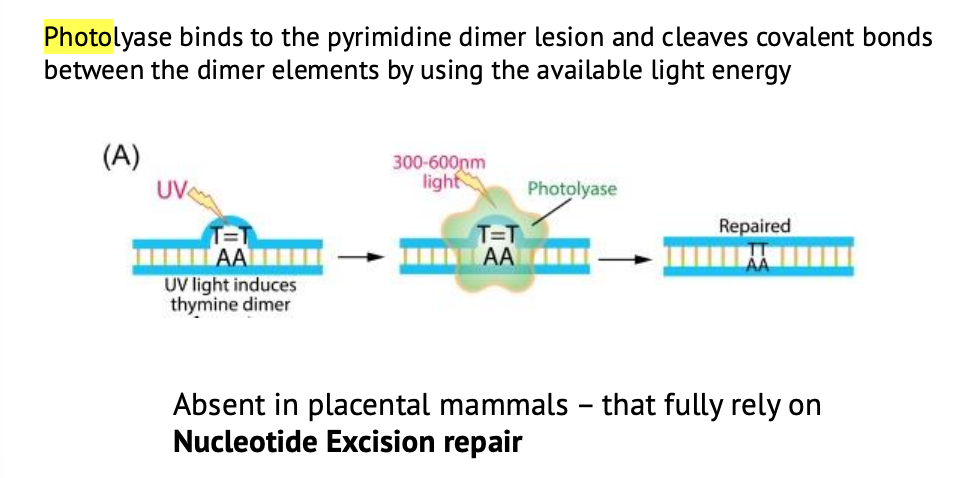
Photoreactivation repair
פתרון אחד הוא photoreactivation בעזרת האנזים photolyase. Photolyase מופעל על ידי אנרגיית אור, ומתקן את החיבור הקוולנטי בין שני התימינים. הוא לא מסיר מקטע DNA שלם ולא מבצע סינתזה מחדש, אלא מפרק ישירות את הקשר הלא תקין בין הבסיסים.
זהו פתרון יעיל מאוד, אבל למרבה הצער הוא לא קיים באדם וביונקים בעלי שליה כמותנו.
ייתכן שהאבות הקדמונים של יונקים בעלי שליה היו פחות חשופים ל־UV, ולכן לא היה לחץ סלקטיבי חזק לשמר את photolyase. מאחר שכל אנזים עולה אנרגטית לתא, מנגנון שאינו מספיק נחוץ עלול להיעלם במהלך האבולוציה. זאת רק השערה.
T4 glycosylase / endonuclease
פתרון נוסף הוא T4 glycosylase / endonuclease, שקיים בבקטריופאג’ים.
המנגנון דומה ברעיון ל־BER: יש אנזים שמזהה T-dimer באופן ספציפי, מסיר או חותך באזור הנזק, וכך מאפשר תיקון.
גם הפתרון הזה לא קיים בבני אדם.
בבני אדם, התיקון המרכזי של T-dimers בפרט ושל bulky lesions בכלל, הוא NER (Nucleotide Excision Repair).
Nucleotide Excision Repair (NER)
NER הוא מנגנון תיקון שמתאים לנזקים גדולים יחסית, כאלה שמשנים את מבנה ההליקס. בניגוד ל־BER, שמסיר בסיס פגוע יחיד, NER מסיר סביב הנזק מקטע DNA.
אפשר לחשוב עליו שוב לפי שלושה שלבים:
- Recognition - זיהוי הנזק
- Processing - פתיחת האזור וחיתוך סביב הנזק
- Repair - השלמת המקטע החסר וסגירת השרשרת
זיהוי הנזק
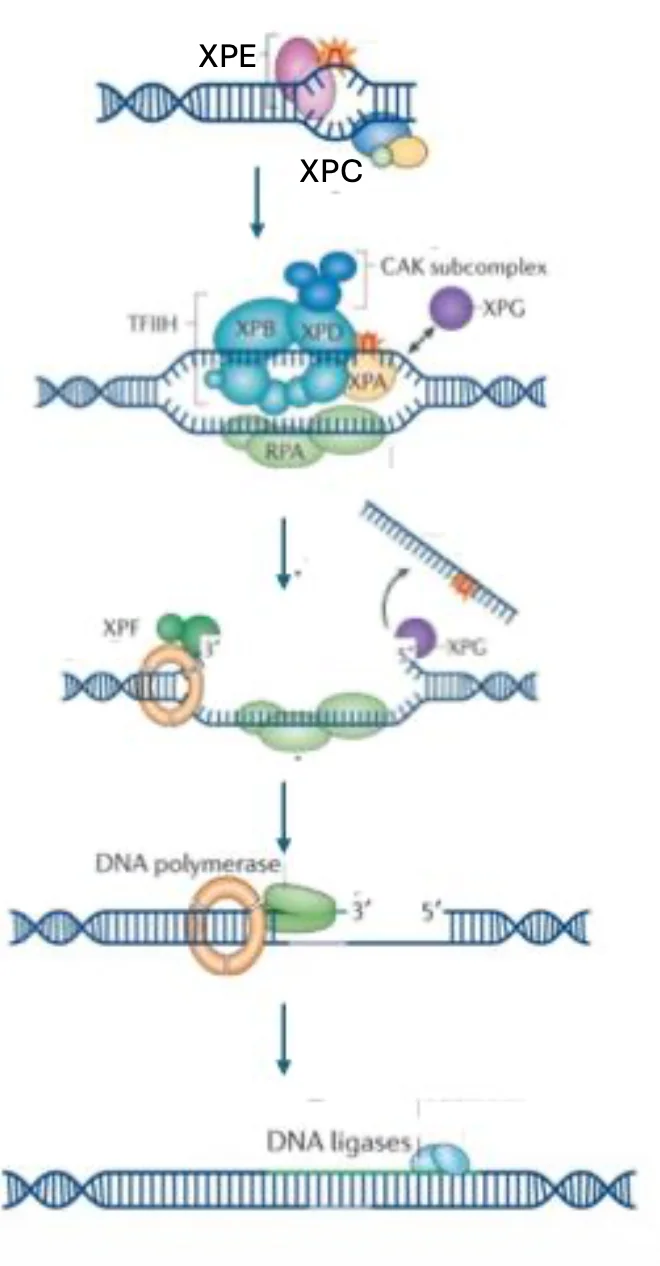
ב־Global Genome NER, הזיהוי נעשה בעיקר על ידי חלבוני XP-C ו־XP-E. הם מזהים מבנה לא תקין של DNA באופן כללי, לא בהכרח ״כימיה״ של בסיס מסוים. הם מתוארים כ־structure-specific proteins: חלבונים שיודעים לזהות עיוות במבנה ה־DNA.
העיוות יכול להיות אזור שבו ההליקס לא תקין, למשל מעבר מקומי למצב פחות דו־גדילי בגלל bulky lesion.
פתיחת האזור
לאחר הזיהוי מגויסים חלבונים נוספים. האזור סביב הנזק צריך להיפתח, ולכן נדרשות הליקאזות:
- XPB
- XPD
הוזכר גם RPA, שתפקידו לשמור על הגדיל החד־גדילי שנפתח, כדי שלא יתפרק או ייסגר בצורה לא רצויה.
חיתוך סביב הנזק
לאחר פתיחת האזור, צריך להסיר מקטע DNA סביב הנזק.
החיתוך נעשה על ידי אנדונוקלאזות:
- XPF
- XPG
המקטע הפגוע מוסר, ואז נשאר פער ב־DNA.
השלמת המקטע
בשלב האחרון:
- פולימראז מסנתז מחדש את המקטע החסר.
- ליגאז מחבר את הקצוות וסוגר את השרשרת.
| שלב | חלבונים מרכזיים | תפקיד |
|---|---|---|
| זיהוי | XP-C, XP-E | זיהוי עיוות מבני ב־DNA |
| פתיחה | XPB, XPD | פתיחת ההליקס סביב הנזק |
| ייצוב | RPA | הגנה על הגדיל החד־גדילי שנפתח |
| חיתוך | XPF, XPG | חיתוך סביב הנזק והסרת המקטע הפגוע |
| תיקון | DNA polymerase, ligase | סינתזה מחדש וסגירת השרשרת |
Xeroderma Pigmentosum (XP)
XP (Xeroderma Pigmentosum) הוא סינדרום גנטי נדיר שנובע מפגיעה בגנים הקשורים ל־NER.
המאפיינים המרכזיים:
- רגישות גבוהה ל־UV
- נטייה מוגברת מאוד לסרטן עור (בסדר גודל של פי 2,000)
- סיכון מוגבר גם לגידולים פנימיים, משום ש־NER אינו מתקן רק T-dimers, אלא גם bulky lesions אחרים שאינם קשורים בהכרח לחשיפה ל־UV.
קבוצות חולים שונות
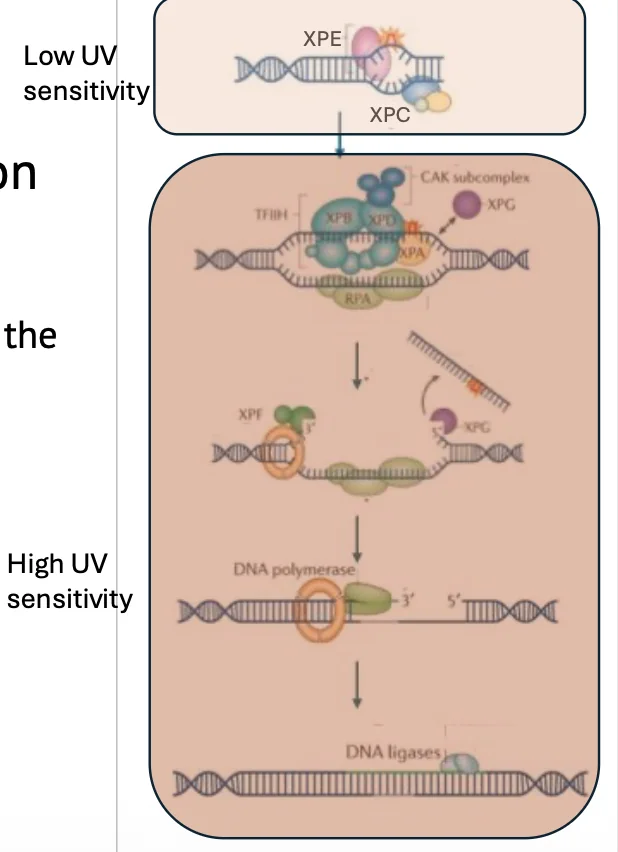
חולי XP מתחלקים לקבוצות לפי הגן שנפגע:
- XPA עד XPG - פגיעה באחד הגנים הקשורים ל־NER.
- XP-V - קבוצה חריגה, שתוסבר בהמשך. אין בה פגיעה בגנים XPA-XPG, אלא במנגנון אחר.
לכל הקבוצות יש נטייה מוגברת לסרטן, אבל יש הבדלים ברגישות ל־UV:
- קבוצות שבהן הפגיעה היא בחלבונים יותר downstream בתהליך, כמו XPA, XPB, XPD, XPF ו־XPG, נוטות לרגישות גבוהה יותר ל־UV.
- לעומת זאת, XP-C ו־XP-E קשורים לזיהוי הראשוני ב־Global Genome NER, ולכן הפגיעה בהם יכולה להיות מעט קלה יותר מבחינת UV sensitivity.
Global Genome NER לעומת Transcription-Coupled NER
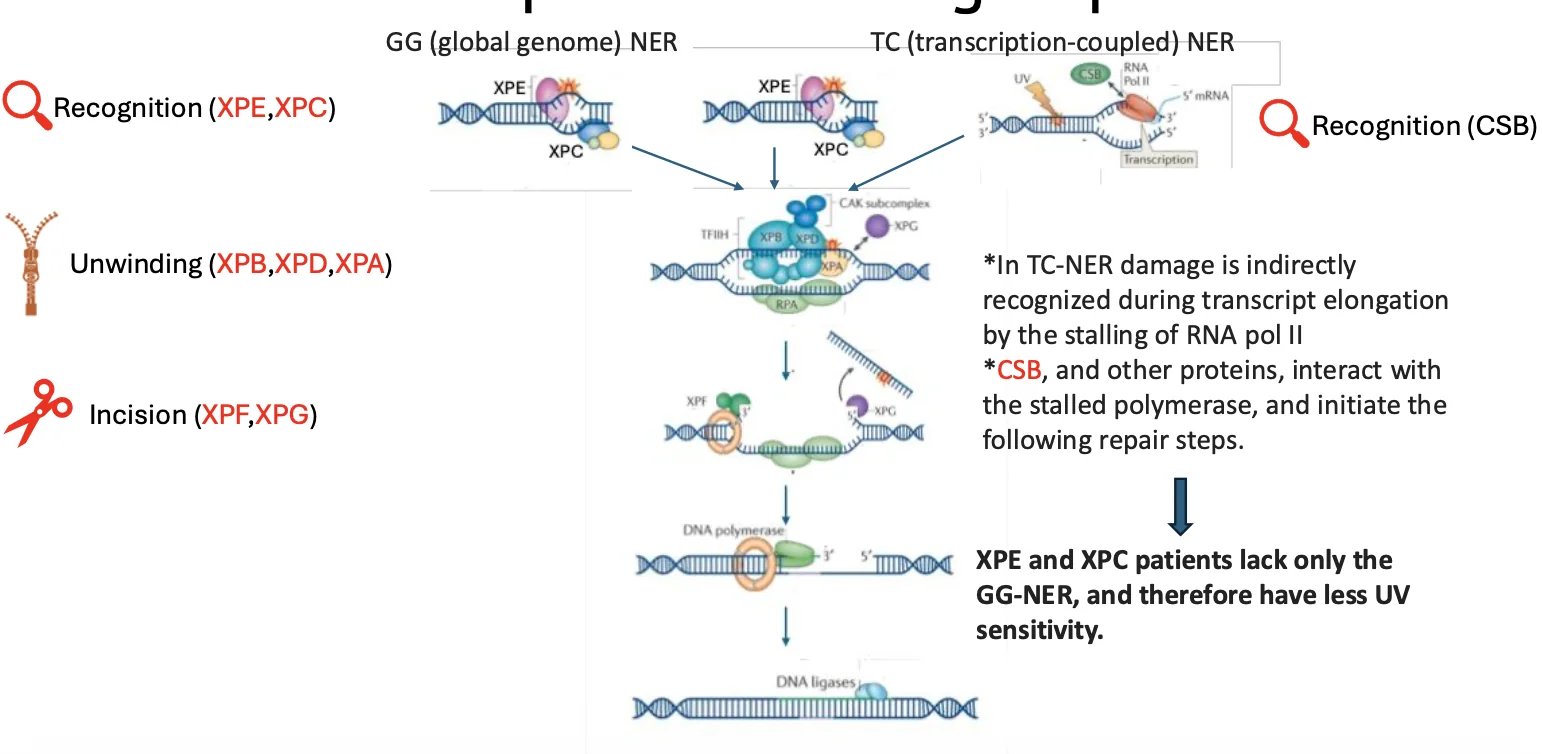
יש שני מסלולים של NER:
- Global Genome NER - GG-NER
- Transcription-Coupled NER - TC-NER
Global Genome NER - GG-NER
המסלול הכללי, שמחפש נזקים ברחבי הגנום. הזיהוי הראשוני בו נעשה על ידי XP-C ו־XP-E.
Transcription-Coupled NER - TC-NER
מסלול שמתמקד באזורים שעוברים שעתוק פעיל. הזיהוי לא מתחיל מחלבון שמזהה את העיוות ב־DNA, אלא ממצב שבו RNA polymerase נתקע על הנזק. כאשר RNA polymerase נעצר, מגויס חלבון כמו CSB, והוא מוביל לגיוס שאר מנגנון התיקון.
הנקודה החשובה: לאחר שלב הזיהוי, שאר המנגנון משותף לשני המסלולים.
לכן:
- אם הפגיעה היא ב־XP-C או XP-E, עדיין נשאר TC-NER באזורים שעוברים שעתוק.
- אם הפגיעה היא בחלבונים downstream, כמו ההליקאזות או האנדונוקלאזות, שני המסלולים נפגעים.
זו הסיבה שבפגיעות ב־XP-C/XP-E, הרגישות ל־UV יכולה להיות קלה יותר יחסית לקבוצות אחרות.
שכפול DNA - כשבעיות קטנות הופכות לגדולות
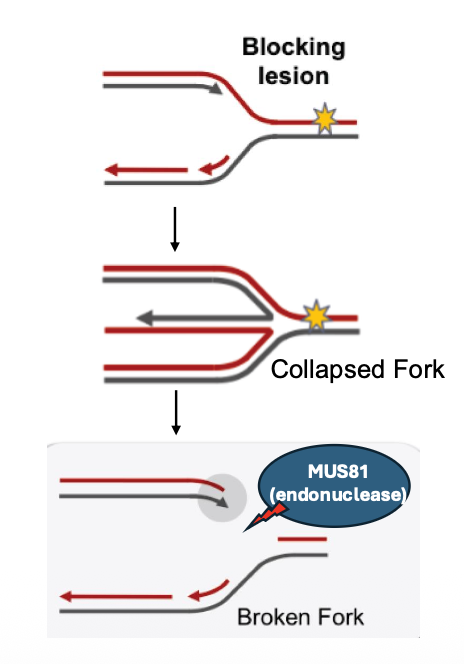
שכפול DNA הוא שלב שבו נזקים קטנים יכולים להפוך לבעיות גדולות. מזלג השכפול צריך DNA תקין כדי להתקדם. כאשר יש bulky lesion, מבנה שניוני, R-loop או מחסור במשאבים כמו dNTPs, הפולימראז יכול להיתקע. מצב כזה נקרא replication stress.
דוגמאות למצבים שיכולים ליצור replication stress:
- מחסור ב־dNTPs
- יותר מדי origins פעילים או התחלת שכפול ביותר מדי אתרים
- מבנים שניוניים ב־DNA
- R-loops
- DNA lesions, במיוחד bulky lesions שלא תוקנו
כשמזלג השכפול נעצר וקורס, נוקלאזות יכולות לזהות את המבנה ולחתוך את ה־DNA. כך נוצרים שברי DNA, כולל שברים דו־גדיליים, שהם מהנזקים המסוכנים ביותר.
לכן התא צריך מנגנונים שמאפשרים לו להמשיך שכפול גם כאשר יש נזק. לא תמיד מדובר בתיקון מלא; לפעמים מדובר בסבילות לנזק.
Translesion Synthesis (TLS)
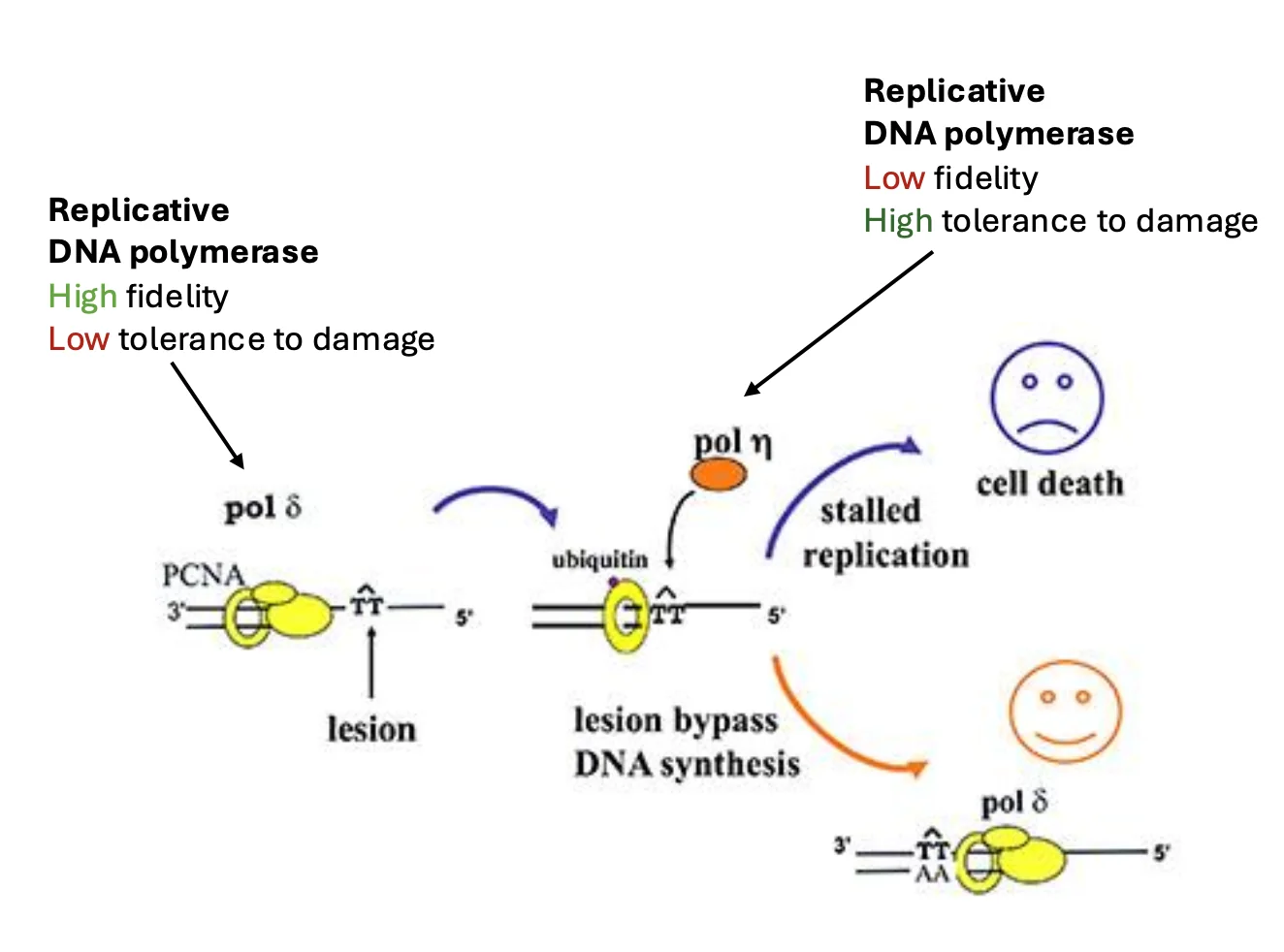
TLS (Translesion synthesis) הוא מנגנון סבילות לנזק, לא מנגנון תיקון קלאסי. המטרה היא לאפשר לשכפול להמשיך למרות שהנזק עדיין נמצא על ה־DNA, לא להסיר את הנזק.
הרעיון:
- הפולימראז הרפליקטיבי נתקע בנזק
- התא מגייס פולימראז TLS
- פולימראז TLS מסוגל לסנתז מעל הנזק
- לאחר מעבר הנזק, הפולימראז הרפליקטיבי יכול לחזור ולהמשיך את השכפול.
היתרון: השכפול ממשיך, ומזלג השכפול לא בהכרח קורס.
החיסרון: פולימראזות TLS פחות מדויקות, ולכן הן יכולות להכניס מוטציות.
למה פולימראזות TLS פחות מדויקות?
פולימראזות רפליקטיביות רגילות מיועדות לדיוק גבוה. הן פועלות טוב כאשר ה־DNA תקין. לעומת זאת, הן מתקשות כאשר יש bulky lesion.
פולימראזות TLS שונות מהן בכמה מאפיינים:
- אין להן proofreading כמו לפולימראזות רפליקטיביות.
- האתר הפעיל שלהן רחב יותר, ולכן הוא יכול להכיל DNA עם נזק גדול או מעוות.
- הן עובדות באופן מהיר ופחות מדויק - פתרון "fast and dirty".
פולימראזות TLS לדוגמה
- Pol η (Polymerase eta): מתמחה בסינתזה מעל T-dimers שנוצרים בעקבות UV. יחסית לפולימראזות TLS אחרות, היא מדויקת יותר בהקשר הזה.
- Pol ι (Polymerase iota): משמשת כגיבוי כאשר Pol η לא פועלת, אבל היא יותר error-prone.
- Pol κ (Polymerase kappa): מתמחה ב־bulky adducts שאינם T-dimers. רמת הדיוק שלה בינונית.
- Pol ζ (Polymerase zeta): משתתפת בהמשך הסינתזה לאחר שפולימראז TLS אחר התחיל לעבור את הנזק.
- Rev1: בעיקר יודע להכניס C, ומשמש בעיקר בהקשרים של AP sites שבהם אין מידע ברור מה צריך להיכנס.
הדגש בשיעור היה בעיקר על Pol η, בגלל הקשר שלה ל־UV ול־XP-V.
מנגנון TLS סביב T-dimer
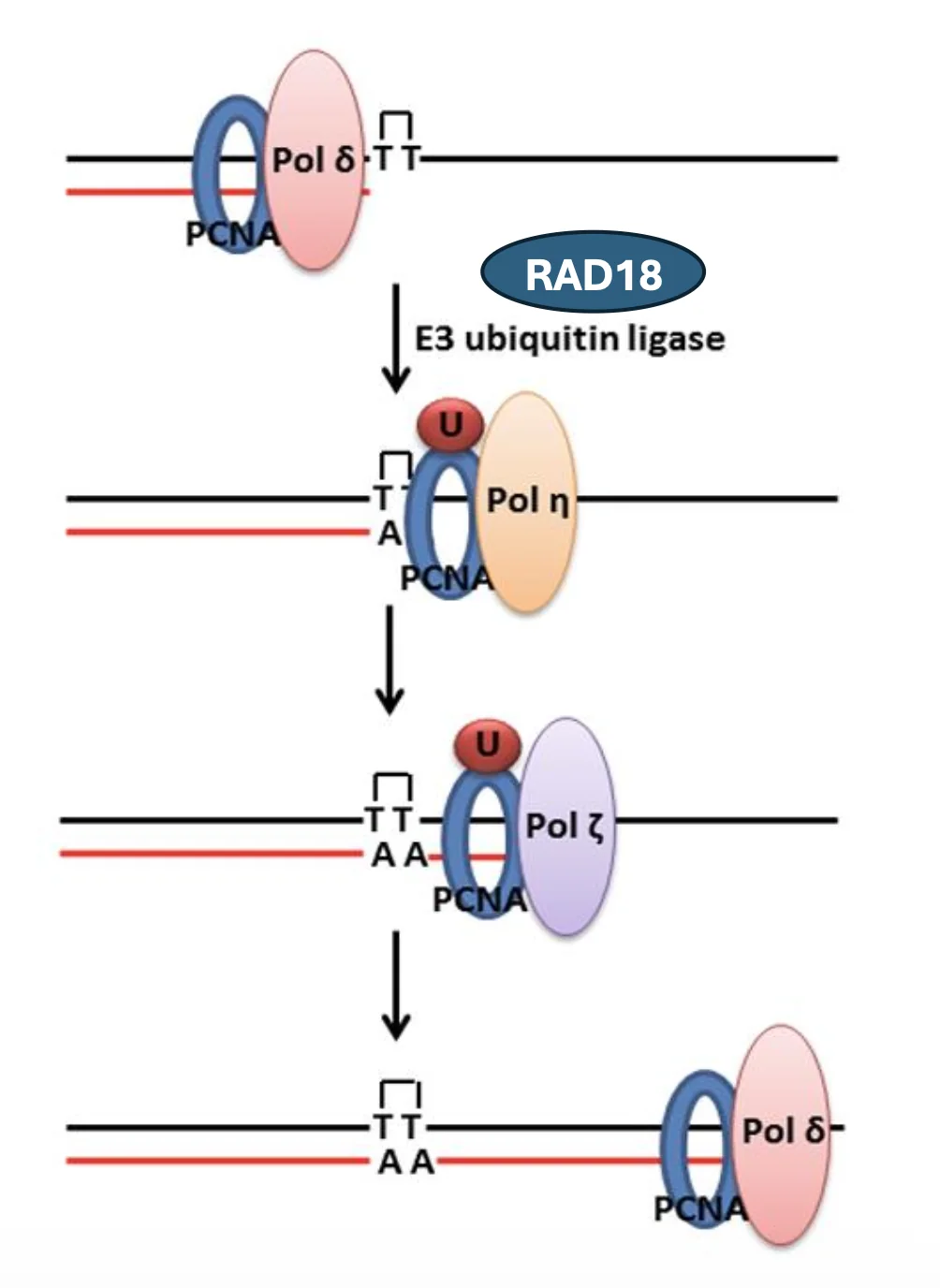
במצב רגיל, פולימראז רפליקטיבי כמו Pol δ מסנתז את ה־DNA יחד עם PCNA.
כאשר Pol δ נתקע ב־T-dimer:
- הפולימראז נעצר.
- PCNA עובר mono-ubiquitination.
- היוביקוויטינציה אינה מסמנת לפירוק, אלא משמשת כסיגנל לגיוס TLS.
- Pol η מגויס לאזור ומסנתז מעל ה־T-dimer.
- Pol ζ ממשיך עוד כמה נוקלאוטידים.
- הפולימראז הרפליקטיבי חוזר ולוקח מחדש את ההובלה.
PCNA מתפקד כאן כמעין פלטפורמה שמקבלת ומעבירה סיגנלים למנגנון השכפול.
XP-V
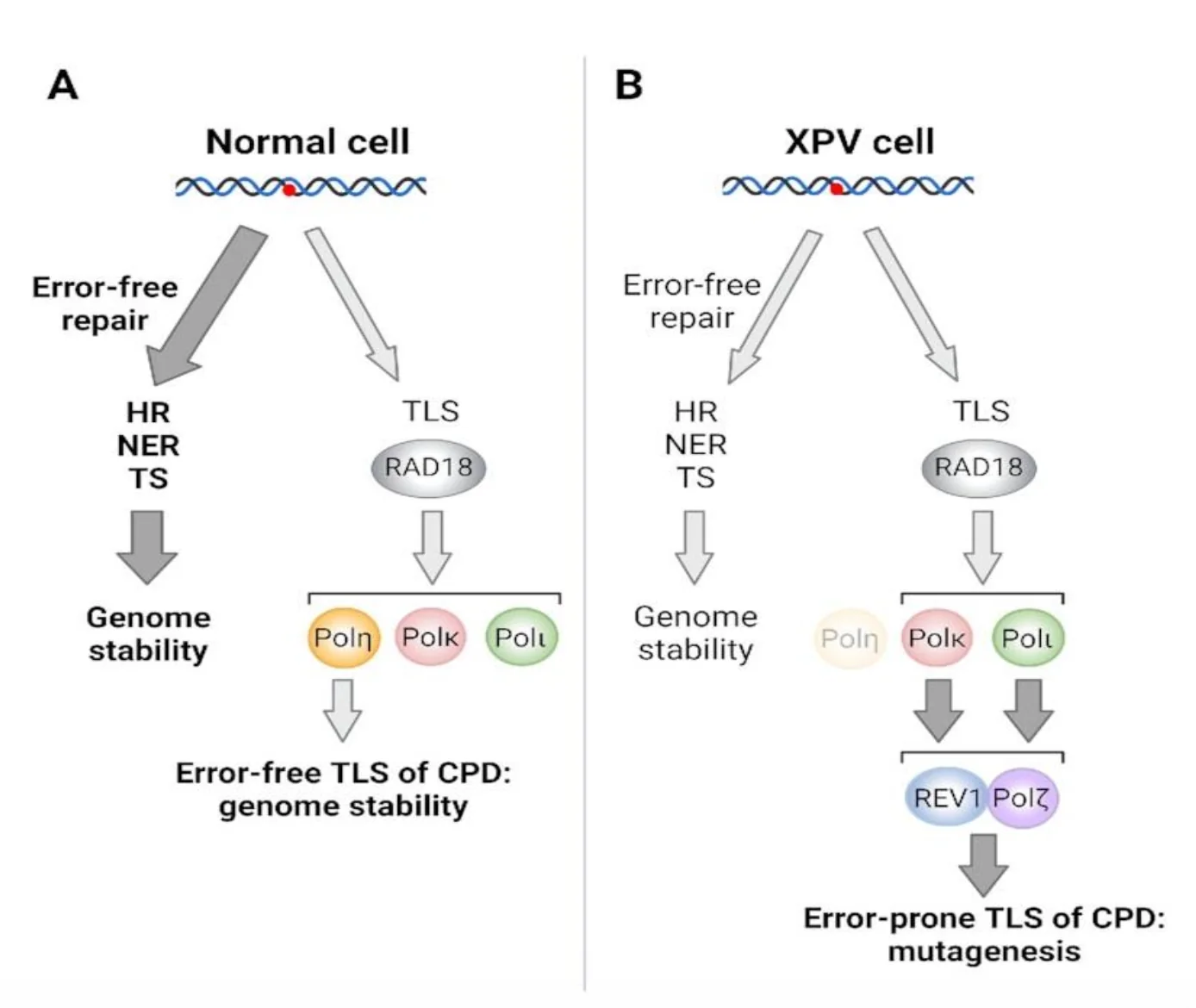
XP-V היא קבוצה חריגה בתוך XP. בחולי XP-V אין פגיעה בגנים XPA-XPG של NER. במקום, יש פגיעה בגן שמקודד ל־Polymerase η.
לכן:
- NER עצמו יכול להיות תקין.
- אבל אם T-dimer לא תוקן לפני השכפול, התא צריך TLS כדי לעבור מעליו.
- בלי Pol η, התא משתמש בפולימראזות TLS אחרות שהן יותר error-prone.
- התוצאה היא יותר טעויות לאחר חשיפה ל־UV ונטייה מוגברת לסרטן עור.
כלומר, XP-V מלמדת ש־NER הוא לא שכבת ההגנה היחידה. גם מנגנוני סבילות לנזק בזמן שכפול חשובים מאוד למניעת מוטציות.
TLS in Cancer
TLS קשור לסרטן בכמה רמות.
חסר ב־TLS תקין
ב־XP-V, חסר Pol η. לכן סינתזה מעל נזקי UV נעשית על ידי פולימראזות פחות מדויקות, ויש יותר מוטציות. זה תורם לנטייה לסרטן עור.
TLS היפראקטיבי (Increased TLS)
מצד שני, גם TLS פעיל מדי יכול להיות בעייתי. אם TLS פועל יותר מדי, התא נוטה להמשיך לשכפל מעל נזקים במקום לעצור, לתקן או לעבור אפופטוזיס.
זה יכול לגרום לשני דברים:
- עלייה במוטציות
- ירידה באפופטוזיס למרות עומס נזק גבוה
בהקשר של טיפול אנטי-סרטני, זה חשוב במיוחד. תרופות כמו cisplatin יוצרות נזקי DNA כדי לעצור שכפול ולהוביל למוות תאי. אם TLS פעיל מדי, התא הסרטני יכול לסבול את הנזק ולהמשיך להתחלק. כך נוצרת סבילות לטיפול.
לכן TLS יכול גם להגן על התא מנזק נקודתי, אבל גם לעזור לתא סרטני לשרוד נזק שהיינו רוצים שיהרוג אותו.
Template Switching
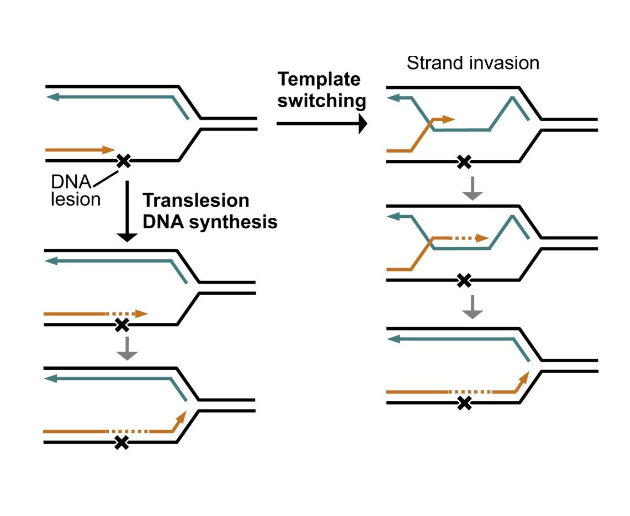
לצד TLS קיים מנגנון נוסף שמאפשר להתמודד עם נזק בזמן שכפול: template switching. במקום לסנתז ישירות מעל הנזק, התא משתמש בגדיל מהכרומטידה האחות כתבנית זמנית.
הרעיון:
- הפולימראז נתקע בנזק על גדיל האב.
- במקום לגייס TLS, אפשר להשתמש ב־DNA שכבר סונתז בכרומטידה האחות.
- לאחר שעוברים את האזור הבעייתי, חוזרים לתבנית המקורית.
זה מנגנון שפועל לצד TLS. הוא דורש זמינות של כרומטידה אחות ולכן רלוונטי בהקשר של שכפול DNA.
Mismatch Repair (MMR)
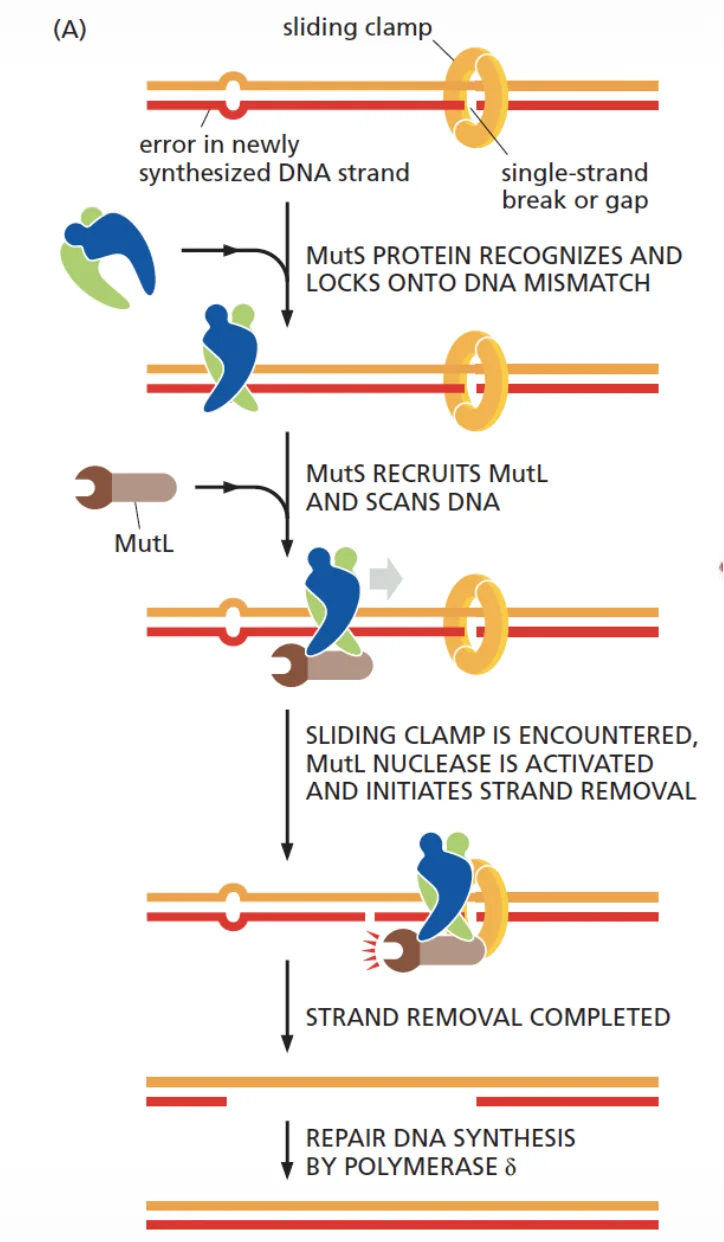
כאן כבר לא מדובר בהכרח ב־bulky lesion או בנזק כימי ברור. לפעמים ה־DNA נראה רגיל מבחינה מבנית, אבל יש בו בסיס לא נכון: mismatch.
Mismatch יכול להיווצר ממספר סיבות:
- פולימראז הכניס בסיס לא נכון בזמן שכפול
- TLS סינתז מעל נזק והכניס בסיס שגוי
- נזק כמו דה־אמינציה או אוקסידציה גרם לזיווג לא תקין
MMR מוריד משמעותית את שיעור הטעויות בגנום.
נתונים:
- פולימראז ללא proofreading מכניס טעות בשיעור: $\sim1:10^5$
- Proofreading מוריד את שיעור הטעויות בעוד כשני סדרי גודל: $\sim1:10^7$
- MMR מוריד את שיעור הטעויות בעוד בערך פי 1,000, עד סדר גודל: $\sim1:10^{10}$
שלבי MMR
כמו במנגנוני תיקון אחרים, גם כאן יש:
- זיהוי
- עיבוד
- תיקון
באופן עקרוני:
- MutS מזהה את ה־mismatch.
- MutL מגויס ומסייע בעיבוד האתר.
- צריך לזהות איזה גדיל הוא החדש ואיזה הוא הישן.
- מסירים את האזור השגוי.
- מסנתזים מחדש את המקטע התקין.
- ליגאז סוגר את השרשרת.
באאוקריוטים השמות שונים, למשל MSH הוא ההומולוג של MutS, אבל העיקרון דומה.
האתגר המרכזי - Strand Discrimination
הבעיה הקשה ב־MMR היא לא רק לזהות שיש mismatch. צריך לדעת איזה גדיל לתקן.
אם יש A מול C, למשל, מי הטעות? האם ה־A נכון וה־C שגוי, או להפך?
התא צריך לזהות את הגדיל החדש, כי שם סביר שהטעות נכנסה בזמן שכפול.
הפתרון של E. coli - מתילציה
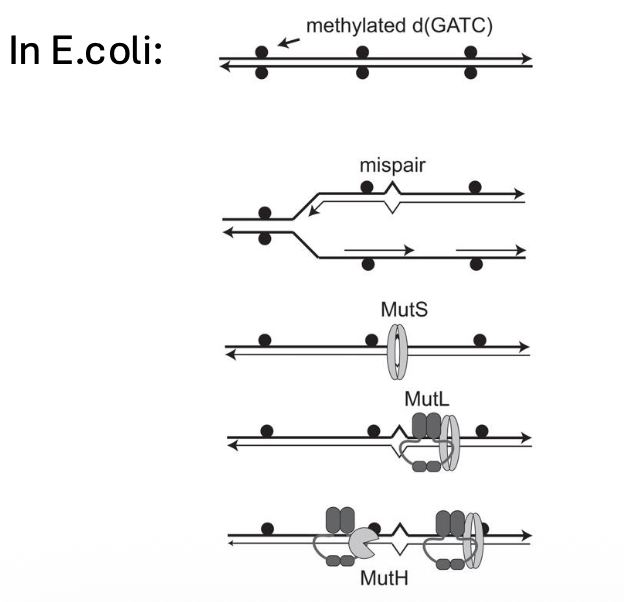
ב־E. coli הפתרון מבוסס על מתילציה של אדנין. מיד לאחר שכפול DNA, הגדיל הישן כבר ממותל, אבל הגדיל החדש עדיין לא הספיק לעבור מתילציה. לכן אפשר לזהות את הגדיל החדש לפי היעדר מתילציה.
העיקרון:
- הגדיל הממותל הוא הישן
- הגדיל הלא ממותל הוא החדש
- מתקנים את הגדיל החדש
הפתרון באאוקריוטים - PCNA וניקים בגדיל החדש
באאוקריוטים אין את המנגנון של מתילציית אדנין לצורך MMR. לכן הזיהוי נעשה אחרת.
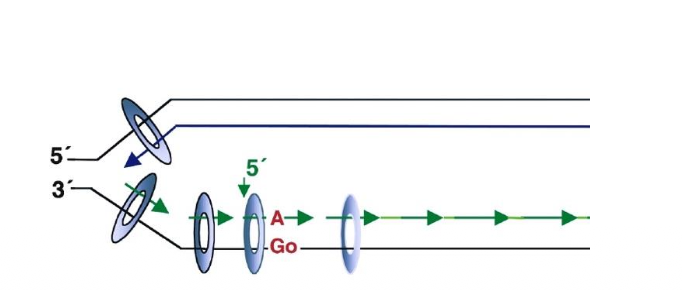
לגדיל החדש יש מאפיינים שמבדילים אותו:
- ב־lagging strand יש מקטעי אוקזקי, ולכן יש ניקים/גאפים זמניים בין המקטעים
- גם ב־leading strand יש קצה חדש באזור מזלג השכפול
PCNA מזהה או מסמן את אזורי הסינתזה החדשה, וכך עוזר לכוון את מנגנון MMR לגדיל החדש.
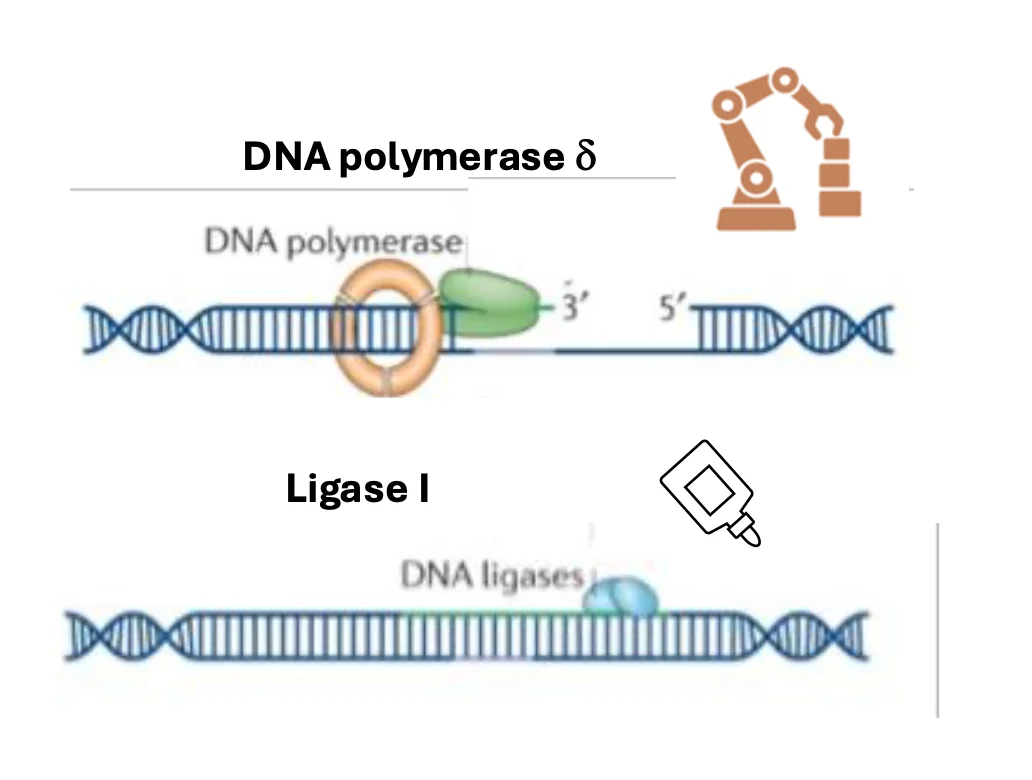
לאחר זיהוי הגדיל הנכון, האזור סביב ה־mismatch מוסר, ואז:
- DNA polymerase δ מסנתז מחדש את המקטע
- Ligase I סוגר את השרשרת
החלק הסופי דומה ברעיון למנגנוני תיקון אחרים: מסירים אזור בעייתי, מסנתזים מחדש, וסוגרים את ה־DNA.
סיכום
בשיעור הקודם התמקדנו בנזקים קטנים ברמת הבסיס. בשיעור הנוכחי עברנו ל־bulky lesions: נזקים גדולים יותר, שנוצרים מחיבור קוולנטי של מולקולות ריאקטיביות או מחיבור בין בסיסים סמוכים, ומשנים את מבנה ההליקס.
הדוגמה המרכזית היא T-dimer בעקבות UV. באורגניזמים מסוימים ניתן לתקן אותו ישירות בעזרת photolyase, אבל באדם התיקון נעשה בעיקר באמצעות NER.
NER מזהה עיוות במבנה ה־DNA, פותח את האזור, חותך סביב הנזק, מסיר מקטע DNA ומסנתז אותו מחדש. פגם בגנים של NER גורם ל־XP, עם רגישות גבוהה ל־UV ונטייה מוגברת לסרטן עור.
אם נזק לא תוקן לפני שכפול DNA, מזלג השכפול עלול להיתקע. כדי לא לקרוס בכל פעם שיש נזק, התא משתמש במנגנוני סבילות כמו TLS. ה־TLS מאפשר סינתזה מעל נזק, אבל במחיר של סיכון מוגבר למוטציות. Pol η חשוב במיוחד לעקיפת T-dimers בצורה יחסית מדויקת; מחסור בו גורם ל־XP-V.
Template switching הוא פתרון נוסף, שבו משתמשים בכרומטידה האחות כתבנית במקום לסנתז ישירות מעל הנזק.
בסוף השיעור עברנו ל־MMR: מנגנון שמתקן טעויות זיווג לאחר שכפול. MMR אינו מתקן bulky lesion עצמו, אלא mismatch שנוצר בזמן שכפול או בעקבות נזק/סינתזה לא מדויקת. האתגר המרכזי בו הוא לזהות איזה גדיל הוא החדש, ובאאוקריוטים PCNA והניקים בגדיל החדש עוזרים לכוון את התיקון.
דור פסקלPCNA משחק תפקיד בשני תהליכים:
- ב־TLS - מגייס את פול אטה
- ב־MMR - Strand discrimination