חלק א’: הפקת DNA - מאיפה מתחילים?
בשביל כל בדיקה גנטית צריך תחילה להפיק DNA, ומסתבר שזאת לא משימה טריוויאלית.
מקור הדגימה - למה זה חשוב?
המקור הנפוץ ביותר לדגימות DNA הוא דם, אבל לא תמיד זאת הבחירה המתאימה: למשל, מטופל שקיבל עירוי דם זמן קצר לפני כן. הדם שנבדק במקרה כזה מכיל גם DNA של התורם, ולא רק של המטופל עצמו, ולכן תוצאות הבדיקה אינן מהימנות.
המסקנה החשובה: תמיד יש לשקול היטב את מקור הדגימה. במטופלים שקיבלו עירוי דם או מוצרי דם, יש לבחור מקור חלופי לדגימת DNA, ולא להסתמך על דם היקפי.
כשרוצים לבדוק גידול
לא תמיד מעוניינים לבדוק את הגנטיקה של המטופל עצמו, אלא דווקא את הגנטיקה של הגידול. תאים סרטניים צוברים עם הזמן מוטציות סומטיות ייחודיות, שהן אלו שמובילות להתפתחות הגידול ולהתנהגותו. לכן, כשמפיקים DNA מדגימת גידול, מתקבל הפרופיל הגנטי של הגידול עצמו, ולא של שאר תאי הגוף של המטופל.
המידע הזה קריטי במיוחד ברפואה מותאמת אישית, משום שהוא מאפשר לזהות מטרות מולקולריות ולקבוע טיפולים ממוקדים שמכוונים למוטציות הספציפיות של הגידול.
איך נראה DNA שהופק?
לאחר הפקה, DNA לא נראה כמו משהו “מעבדתי” קלאסי. בפועל, מדובר בחומר לבן-חלבי, צמיג וסיבי, שלעיתים ניתן לראותו בעין בלתי מזוינת: חומר סמיך ועשיר ב־DNA, בדומה לחומר ביולוגי שמכיל תאים מפורקים וריכוז גבוה של חומצות גרעין.
ההשוואה נועדה להדגיש נקודה חשובה: כשתאים מתפרקים ומשתחרר מהם DNA בכמות גדולה, מתקבל חומר בעל מרקם צמיגי וסיבי (״נזלת״), שהוא מאפיין אופייני של DNA לאחר הפקה מוצלחת.
חלק ב׳: PCR - הבדיקה הבסיסית ביותר בגנטיקה
רקע היסטורי
PCR (Polymerase Chain Reaction) פותחה בשנות ה־90 וזיכתה את מפתחיה בפרס נובל. זאת הבדיקה הבסיסית ביותר בגנטיקה, ובלעדיה מרבית הגנטיקה המעבדתית המודרנית לא הייתה מתאפשרת.
העיקרון: הגברה של מקטע מוגדר
הגנום האנושי כולל מיליארדי בסיסים, אך לעיתים נדרש ניתוח של אזור קטן ומדויק בלבד - למשל, ניתוח של אקסון ספציפי בגן מסוים. PCR מאפשרת הגברה ממוקדת של אותו מקטע, באמצעות יצירת מיליוני עותקים שלו, לצורך המשך עבודה אנליטית.
כיצד זה עובד?
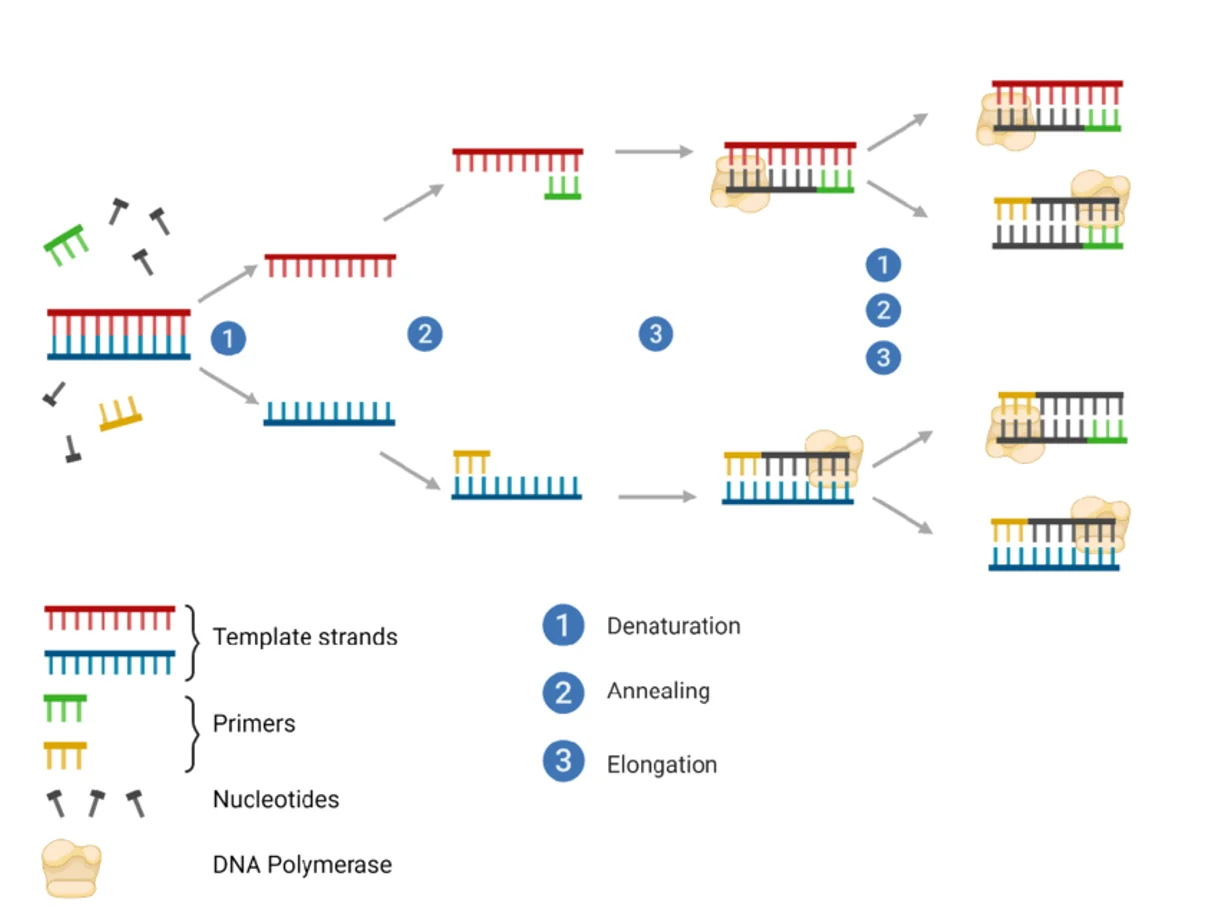
התהליך דומה מאוד לשכפול DNA טבעי, ומורכב משלושה שלבים מחזוריים:
Denaturation- חימום ה־DNA לצורך הפרדת שני הגדיליםAnnealing- קירור המאפשר היצמדות של primers לרצף המטרהExtension- Taq polymerase מאריך את הגדילים החדשים
המחזורים חוזרים מספר רב של פעמים, ובכל מחזור כמות המקטע מוכפלת. לאחר כ־30 מחזורים מתקבלים מיליוני עותקים של המקטע המבוקש.
Primers - רכיב מפתח ב־PCR
שכפול DNA ללא primer אינו אפשרי: ה־polymerase אינו מתחיל סינתזה מאפס, אלא דורש רצף קצר שמשמש בתור נקודת התחלה.
מה שייחוד ב־PCR הוא שה־primers מתוכננים מראש בהתאם לאזור הרצוי להגברה: נקבע מקטע המטרה, ונבחרים primers הנקשרים אליו באופן ספציפי.
דרישות ל־primer איכותי
- דיוק - התאמה מלאה לרצף המטרה.
- ייחודיות - primer קצר מדי (למשל ATG) יופיע במקומות רבים בגנום ולכן עלול להיקשר לאתרים רבים. נדרש primer ארוך מספיק כדי להיות ייחודי סטטיסטית.
- שני כיוונים - נדרשים Forward primer ו־Reverse primer, אחד מכל צד של המקטע. כך מוגבר האזור שביניהם, ובמקרים מסוימים מתאפשר גם ריצוף משני הכיוונים.
בעיה קלאסית: Pseudogenes
בבדיקות מסוימות קיימת מגבלה משמעותית עקב pseudogenes - רצפים בגנום שדומים לגן האמיתי אך אינם פונקציונליים. primer עלול להיקשר ל־pseudogene במקום לגן האמיתי. במצב כזה תתקבל הגברה של רצף שאינו המטרה, דבר שעלול להוביל לתוצאות שגויות.
חלק ג׳: עבודה עם RNA
למה בכלל RNA אם ה־DNA זהה בכל תא?
נכון - ה־DNA זהה בכל תאי הגוף. הסיבה לעבוד עם RNA היא ש־RNA משקף ביטוי גנים. לא כל גן פעיל בכל תא: תא לב יבטא גנים של חלבוני לב, ותא מוח יבטא גנים הקשורים לנוירוטרנסמיטורים. לכן, כשנדרש לבדוק אם גן מסוים מתבטא ברקמה מסוימת, המידע הרלוונטי נמצא ב־RNA ולא ב־DNA.
מתי בודקים RNA?
- בדיקת ביטוי גנים - האם גן מסוים פעיל ברקמה מסוימת.
- הערכת המשמעות של מוטציות ב־splicing - כאשר קיים שינוי באזור של splice site ורוצים לדעת האם הוא אכן משפיע על תהליך ה־splicing, יש לבדוק את ה־RNA ולראות מה התקבל בפועל.
האתגר: RNA אינו יציב
בניגוד ל־DNA שיכול להישמר לאורך זמן, RNA מתפרק במהירות ובעל זמן מחצית חיים קצר. לכן נדרשת הפקה ועבודה מהירה ומבוקרת.
הפתרון: cDNA
כדי לעבוד עם RNA בצורה נוחה, מבצעים המרה שלו ל־DNA באמצעות האנזים Reverse Transcriptase (מוכר מווירוסים כמו HIV).
תהליך העבודה:
mRNA → Reverse Transcription → output: cDNA (complementary DNA)
המאפיין המרכזי של cDNA הוא שהוא לא מכיל אינטרונים, משום שה־mRNA כבר עבר splicing. לכן cDNA מייצג בפועל את ״הגן בלי האינטרונים״.
בשלב הזה ניתן להגביר את ה־cDNA באמצעות PCR רגיל ולהמשיך בעבודה מעבדתית.
חלק ד׳: Gel Electrophoresis - הפרדה לפי גודל
העיקרון
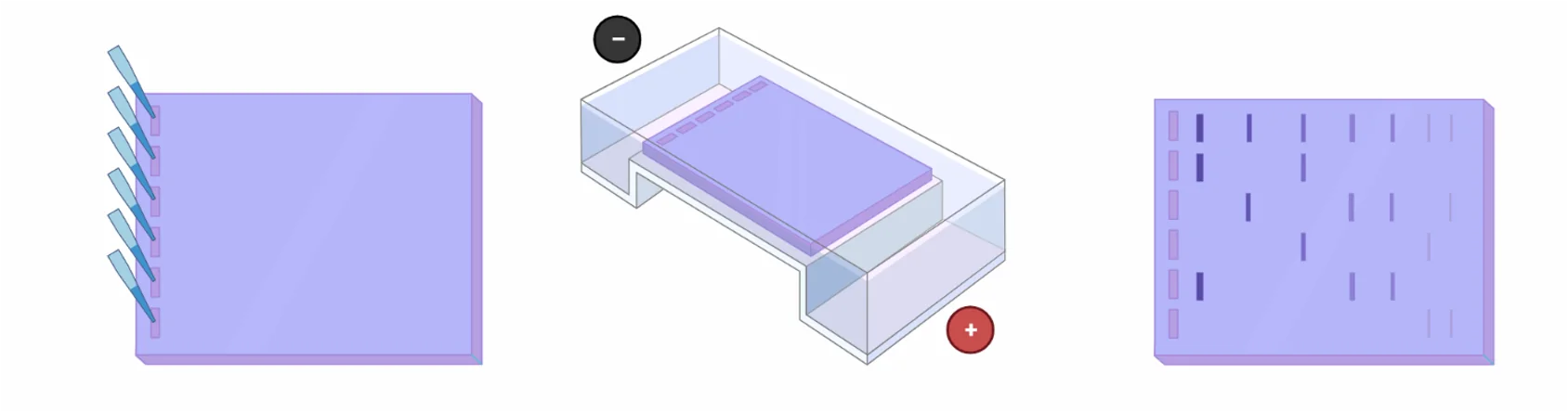
ג׳ל אלקטרופורזה מאפשר הפרדה של מולקולות לפי גודל ומטען. את הדגימה טוענים על ג׳ל, מפעילים שדה חשמלי, והמולקולות נעות דרכו.
- מולקולות קטנות נעות מהר יותר בג׳ל
- מולקולות גדולות נעות לאט יותר
בסיום מתקבלת הפרדה: כל מקטע מופיע במיקום שונה בהתאם לגודלו.
למה זה שימושי?
לאחר PCR ניתן להריץ את התוצר על ג׳ל כדי לבדוק:
- האם בכלל התקבל תוצר
- מה הגודל של התוצר שהתקבל
- האם הגודל שהתקבל תואם את הגודל הצפוי
כמעט תמיד משתמשים גם ב־Ladder (סולם) - תערובת של מקטעים בגדלים ידועים - לצורך השוואה והערכת גדלים.
חלק ה׳: שיטות Blotting - Western, Northern, Southern
הרעיון הכללי
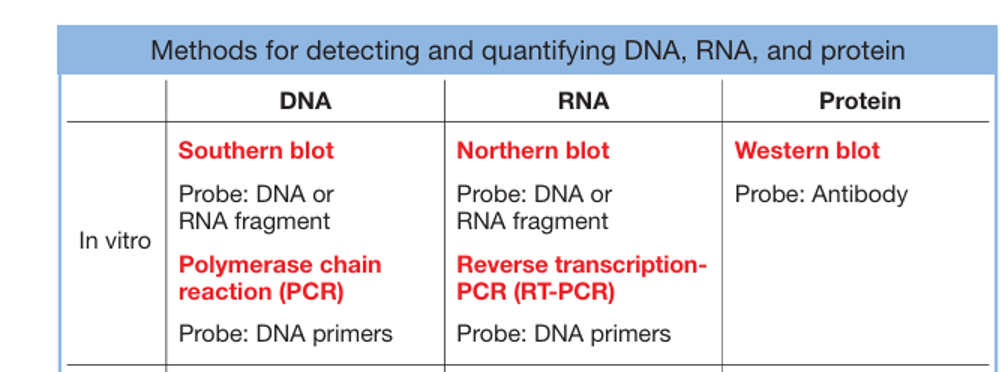
שיטות ה־Blotting פועלות על עיקרון משותף: הפרדה לפי גודל, העברה לממברנה, ולאחר מכן זיהוי של מולקולה ספציפית באמצעות נוגדן או probe.
שלושת הסוגים
| שיטה | מה נבדק |
|---|---|
| Western Blot | חלבונים |
| Northern Blot | RNA |
| Southern Blot | DNA |
השם Southern נקבע על שם החוקר Edwin Southern. בהמשך, כשפותחו שיטות מקבילות ל־RNA ולחלבונים, נקבעו השמות Northern ו־Western כבדיחה “גיאוגרפית”.
כיצד Western Blot עובד?
- הפקה - הפקת חלבונים מהתאים
- הפרדה - הרצה על ג׳ל; החלבונים נפרדים לפי גודל
- העברה לממברנה - העברת החלבונים מהג׳ל לממברנה ייעודית
- נוגדן ראשוני - הוספת נוגדן ספציפי לחלבון הנבדק
- נוגדן שניוני - הוספת נוגדן שמזהה את הנוגדן הראשוני, והוא פלואורסצנטי
- קריאה - זיהוי פס זוהר במקום שבו נמצא החלבון
למה נדרשים שני נוגדנים?
הנוגדן הראשוני מספק את הספציפיות לחלבון המטרה, אבל הוא לא מסומן ולכן אינו “נראה” ישירות. הנוגדן השניוני נקשר לנוגדן הראשוני (כלומר, “נוגדן נגד נוגדנים”) ומסומן כך שניתן לזהות את מיקום החלבון על הממברנה.
חשיבות ה־Control
תמיד נדרש Control.
מקובל להשתמש ב־Housekeeping gene/protein - גן או חלבון שמצויים באופן קבוע בתאים, כמו Actin או GAPDH. מטרת הבקרה היא להבחין בין היעדר ביטוי אמיתי לבין כשל טכני (הפקה לא תקינה, נוגדן שלא עבד, העברה לא טובה וכדומה). כאשר גם ה־housekeeping לא מופיע, הסבירות הגבוהה היא לתקלה טכנית ולא למסקנה ביולוגית.
בדוגמה שהוצגה בהרצאה נבדק אינסולין בתאי כליה לעומת תאי לבלב: בבקרת housekeeping (למשל RPS7) הופיע פס בשני סוגי התאים, בעוד שבאינסולין הופיע פס רק בלבלב ולא בכליה - תוצאה הגיונית, משום שאינסולין מיוצר בלבלב ולא בכליה.
מה עדיין בשימוש כיום?
- Western Blot - עדיין בשימוש נרחב, בעיקר במחקר, לזיהוי ביטוי של חלבון ספציפי.
- Northern Blot - פחות נפוץ, משום שקיימות שיטות יעילות יותר לבדיקת RNA.
- Southern Blot - כמעט ואינו בשימוש, למעט בדיקה אחת: Fragile X syndrome. במחלה זו קיים אזור של חזרות DNA שמתארך, וריצוף אזורים חזרתיים עלול להיות מאתגר. Southern blot מאפשר להעריך את גודל האזור ובכך מסייע באבחנה.
חלק ו׳: Restriction Enzyme Analysis - זיהוי מוטציות ידועות (RELP)
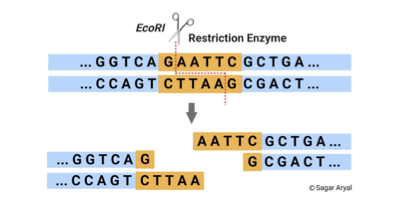
העיקרון - חיתוך DNA ברצפים ספציפיים
אנזימי Restriction (Restriction Enzymes) חותכים DNA ברצפים ספציפיים. לדוגמה, אנזים מסוים יחתוך בכל פעם שיזהה את הרצף GAATTC. כאשר קיימת מוטציה שמשנה את הרצף הזה, ייתכן שהאנזים כבר לא יזהה את האתר - ולכן לא יתרחש חיתוך.
כאשר קיימת מוטציה ידועה (למשל, שינוי G ל־C בעמדה מסוימת), ניתן להבדיל בין רצף תקין לבין רצף מוטנטי לפי דפוס החיתוך. לדוגמה:
- רצף תקין: G|AATTC (נחתך)
- רצף מוטנטי: CAATTC (לא נחתך)
תהליך העבודה
- הגברת האזור ב־PCR
- הוספת האנזים החותך
- הרצה על ג׳ל
תוצאות אפשריות
- נחתך ← מתקבלים שני פסים קטנים
- לא נחתך ← מתקבל פס אחד גדול
- הטרוזיגוט ← מתקבלים שלושה פסים (אלל אחד נחתך ואלל אחד לא)
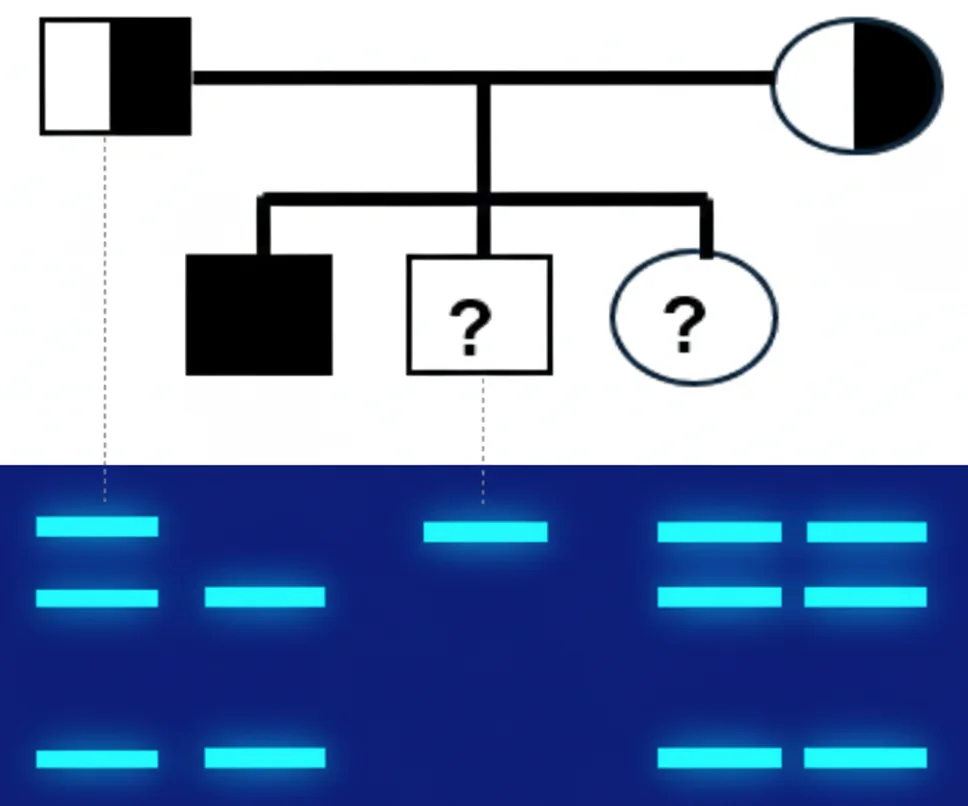
דוגמה קלינית שהוצגה בהרצאה
משפחה עם מחלה רצסיבית:
- ההורים (נשאים) - שלושה פסים: אלל אחד עם מוטציה (לא נחתך) ואלל אחד תקין (נחתך)
- הילד החולה - פס אחד: שני האללים עם מוטציה (לא נחתכים)
מתי משתמשים ב־Restriction Enzyme Analysis?
השיטה מתאימה רק כאשר המוטציה ידועה מראש; לא ניתן לגלות בעזרתה מוטציות חדשות.
שימוש נפוץ הוא Population Screening: כאשר מזוהה מוטציה חדשה באוכלוסייה/יישוב מסוים ונדרש להעריך שכיחות. ריצוף מאות דגימות הוא מאמץ גדול, בעוד ש־PCR + אנזים חיתוך + ג׳ל עבור מספר רב של דגימות הוא תהליך פשוט יותר, זול ומהיר.
נקודה חשובה: תלוי בתכנון
ייתכן מצב שבו המוטציה נחתכת והתקין לא, או להפך - הכול תלוי ברצף הספציפי ובאנזים שנבחר. בשאלות מבחן המידע הדרוש לכך יינתן במפורש.
דוגמה משיעורי הבית
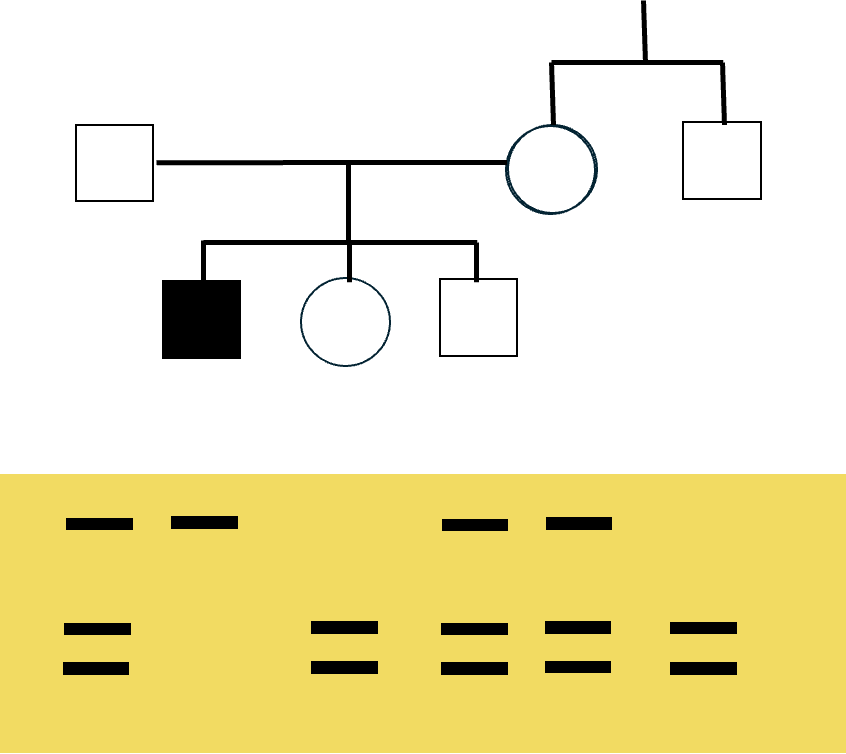
- האבא (שמאל מעלה) - שלושה פסים מכאן שהוא נשא כי אלל אחד נחתך והשני לא. נסמן N אלל תקין ו m מוטנטי, אז הגנוטיפ של האב יהיה N/m. ההסבר הוא פשוט ששני פסים מייצגים אתר חיתוך, אז יש אתר חיתוך ועוד אלל נוסף.
- הבן החולה (שמאל למטה) - אין אתר חיתוך אז פס אחד, כלומר m/m (האלל לא נחתך). חולה רצסיבי.
- הבת - לא נשאית. יש שני פסים אז N/N כי האתר נחתך ומכאן שיש אתר חיתוך.
- הבן הימני (עם שלושת הפסים) - דומה לאבא, הוא נשא עם גנוטיפ של N/m כמפורט לעיל.
- האמא - גם כן נשאית, מאותם שיקולים הגנוטיפ שלה יהיה N/m.
- הדוד (עם שני פסים) - בדומה לבת הגנוטיפ יהיה N/N, הוא לא נשא.
חלק ז׳: Sanger Sequencing - הקלאסיקה
רקע
Sanger Sequencing הייתה שיטת הריצוף המרכזית לגנים לפני עידן ה־NGS. השיטה פותחה בשנות ה־70 על ידי פרדריק סנגר וזיכתה אותו בפרס נובל. גם Human Genome Project השלים את ריצוף הגנום האנושי באמצעות Sanger - פרויקט עצום שנמשך שנים.
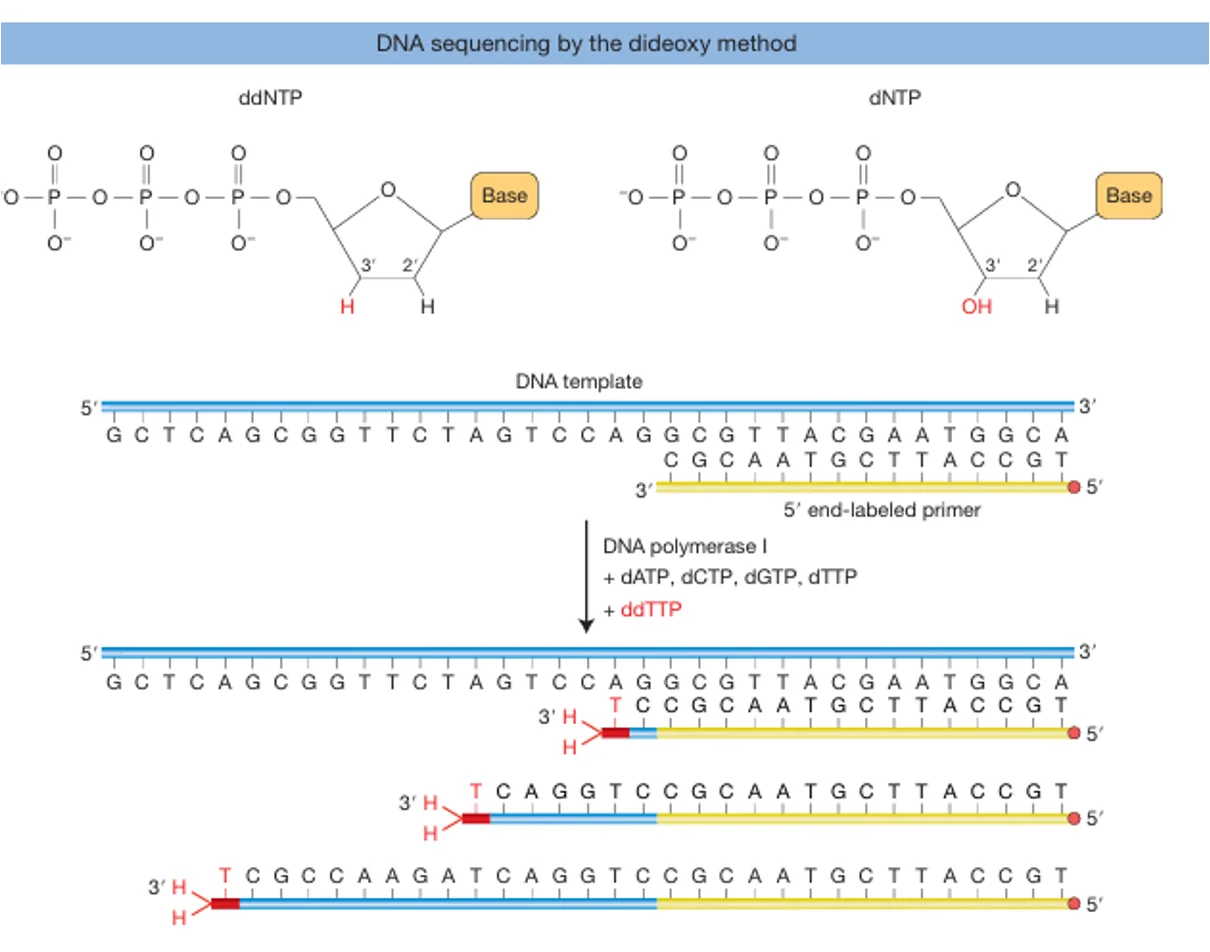
העיקרון הגאוני
בריאקציית PCR רגילה משתמשים בנוקלאוטידים תקינים (dNTPs) שמאפשרים לשרשרת להמשיך להתארך. לעומת זאת, בשיטת Sanger מוסיפים גם ddNTPs (dideoxy nucleotides) - נוקלאוטידים “פגומים” שחסרה בהם קבוצת OH. כאשר ddNTP משתלב בשרשרת, ההתארכות נעצרת ולא ניתן להוסיף אחריו נוקלאוטידים נוספים.
מה מתרחש בפועל?
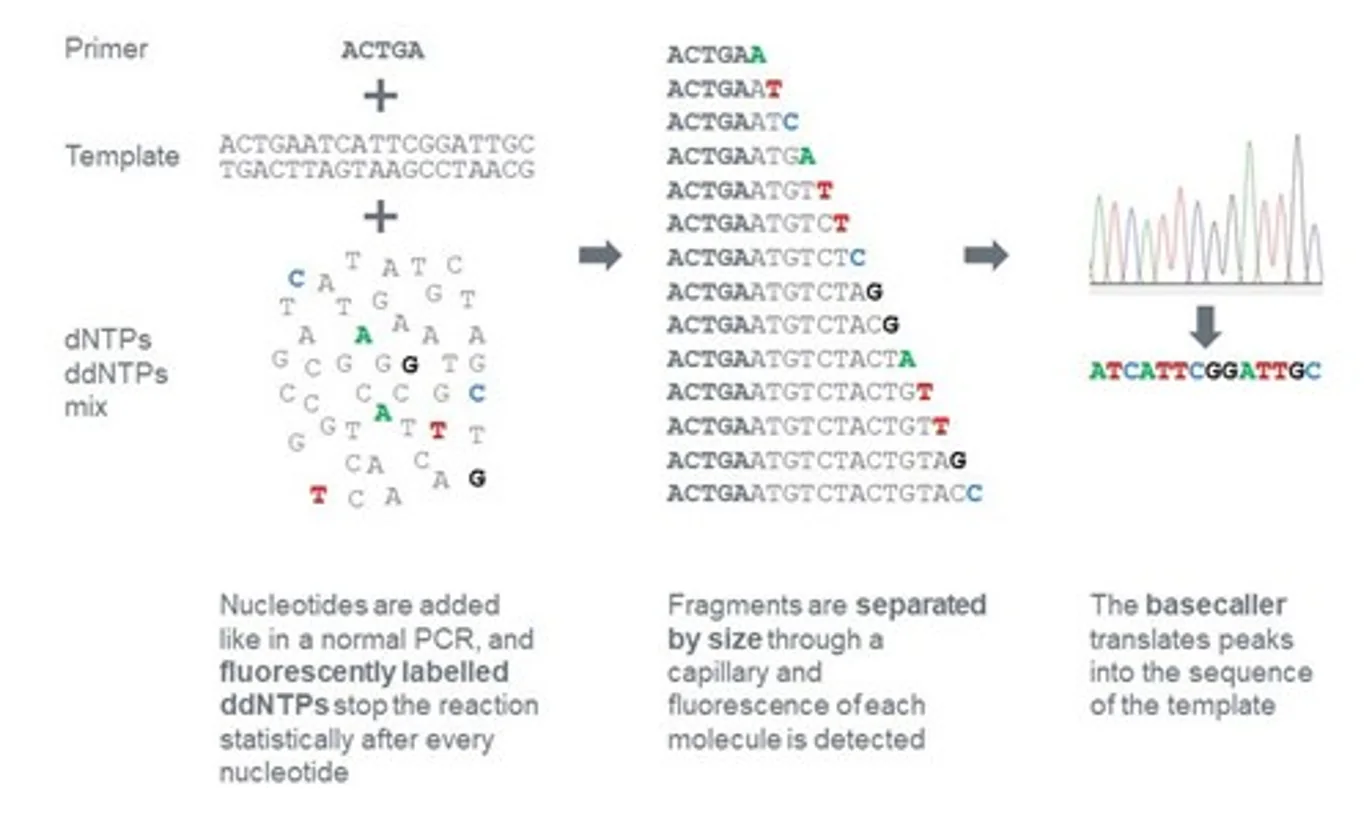
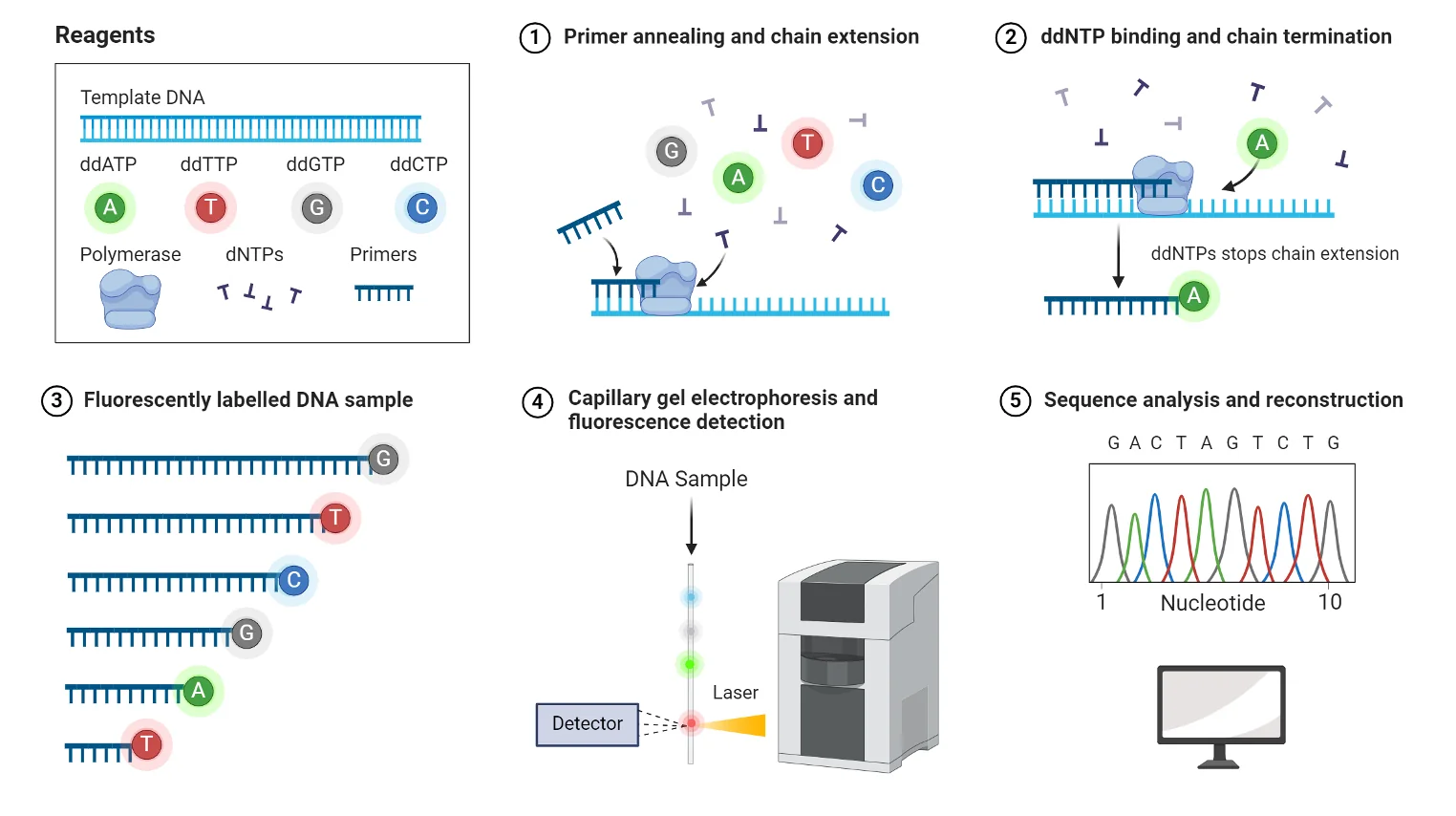
מערבבים dNTPs עם כמות קטנה של ddNTPs. באופן אקראי, לעיתים ddNTP נכנס לשרשרת ועוצר אותה. לאחר ביצוע הריאקציה פעמים רבות מתקבל אוסף של מקטעים בכל האורכים האפשריים, משום שהעצירה מתרחשת כל פעם בעמדה אחרת.
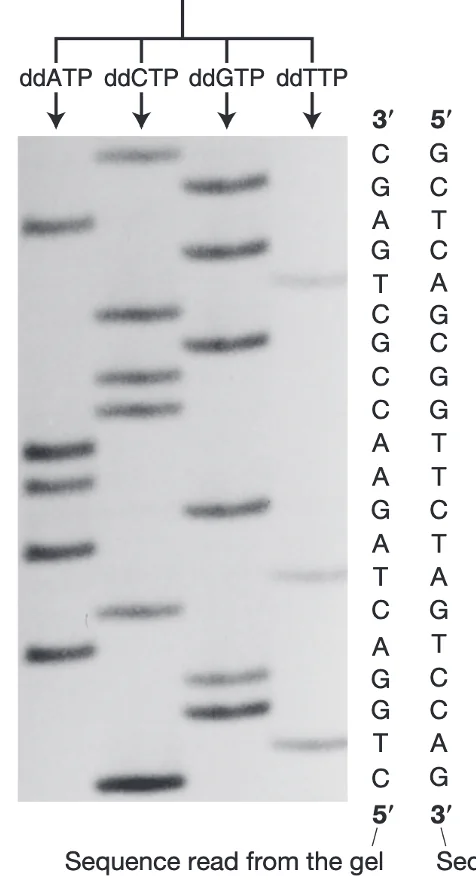
איך מתקבל הרצף?
כל אחד מארבעת ה־ddNTPs (A, T, C, G) מסומן בצבע שונה. את תוצרי הריצוף מריצים על ג׳ל (או בקפילרה), שם הם נפרדים לפי גודל. עבור כל אורך מזוהה הצבע הזוהר, והוא קובע מהו הבסיס באותה עמדה.
 |  |
דגש: בריצוף בשיטת סנגר מתחילים מריאקציית PCR, כלומר, מרצפים קטע קטן יחסית של דנ״א (עד 1000 בסיסים) מתוך מקטע דנ״א שהוגבר.
הוזכר בהרצאה שבעבר הקריאה נעשתה ידנית מתוך הג׳ל (“כאן A, כאן T…”), אבל כיום התהליך מתבצע באופן אוטומטי (למעט בשיעורי הבית שקיבלנו).
מגבלות
- אורך מוגבל - ניתן לרצף בערך כ־500 בסיסים בריאקציה אחת; מעבר לכך איכות הקריאה יורדת.
- עבודה ידנית - נדרש תכנון primers לכל אקסון, ביצוע PCR וריצוף. גן עם 72 אקסונים (כמו NF1) משמעותו לפחות 72 ריאקציות נפרדות.
- זמן ועלות - ריצוף גן שלם דורש זמן רב.
מתי עדיין משתמשים ב־Sanger?
- מוטציות ידועות - כאשר ידוע בדיוק מה מחפשים, Sanger ממוקד יכול להיות מהיר וזול יותר מ־NGS.
- אישור ממצאים - לעיתים מאמתים ממצאים שהתקבלו ב־NGS באמצעות Sanger.
- בדיקות משפחתיות - לאחר זיהוי מוטציה במשפחה, ניתן לבדוק קרובים בצורה נוחה באמצעות Sanger.
דוגמה משיעורי הבית: פיענוח תוצאות ריצוף בג׳ל

הרצף שהתקבל בג׳ל:
- 3’- AGATTTCCAGGTCCCCAGTGGAAAGCTT -5’
- 5’ - TCTAAAGGTCCAGGGGTCACCTTTCGAA -3’
דוגמה נוספת:
האחות נשאית לשינוי והאח אינו נשא.
חלק ח׳: Next Generation Sequencing - המהפכה
מה נשתנה?
במקום לרצף מקטע אחד בכל פעם, NGS מאפשר ריצוף של מיליוני מקטעים במקביל - שינוי שהפך את התחום כולו.
העיקרון - ארבעה שלבים מרכזיים
- שבירה - חיתוך ה־DNA למקטעים קצרים
- Capture - שימוש ב־probes (כמו הרבה primers) כדי “לתפוס” את האזורים המיועדים לריצוף
- ריצוף מקבילי - המכשיר מרצף את כל המקטעים בו־זמנית
- Alignment - המחשב מסדר את המקטעים לפי הסדר הנכון, בדומה להרכבת פאזל
Alignment - סידור הפאזל
לאחר הריצוף מתקבלים מיליוני מקטעים קצרים. כדי לדעת מאיפה כל מקטע הגיע, משתמשים ב־Reference sequence - רצף ייחוס מוכר של הגנום האנושי (מפרויקט Human Genome וממאגרי מידע גדולים של אנשים בריאים).
האלגוריתם משווה כל מקטע לרצף הייחוס ומאתר את המיקום המתאים לו - כמו התאמת חתיכת פאזל לתמונה השלמה.
חפיפה מסייעת: המקטעים אינם זהים באורכם, ולעיתים קיימת חפיפה בין מקטעים סמוכים. החפיפה מחזקת את הוודאות שהסידור נכון.
Coverage ו־Depth - מדדי איכות
- Coverage - האם כל האזור הרצוי אכן רוצף. ייתכן שאזור מסוים לא “נתפס” על ידי ה־probes או שהוא בעייתי מבחינה כימית. אזור שאינו מכוסה אינו מאפשר הסקת מסקנות לגביו.
- Depth - כמה פעמים כל עמדה רוצפה. קריאה בודדת יכולה להיות טעות; לעומת זאת, קריאה חוזרת (למשל 30 פעמים) מעלה משמעותית את רמת הביטחון.
הוזכר בהרצאה כי הסטנדרט המינימלי הוא לפחות 20 קריאות כדי לקבוע בביטחון את הבסיס בעמדה מסוימת.
כיצד מזהים הטרוזיגוטיות?
אם בעמדה מסוימת מתקבלות למשל 50% קריאות של T ו־50% קריאות של C, הדבר מתאים להטרוזיגוטיות: אלל אחד עם T ואלל אחד עם C. אם מתקבלות 100% קריאות של T, מדובר בהומוזיגוטיות.
סוגי בדיקות NGS
- Panel - ריצוף רשימת גנים מוגדרת מראש. לדוגמה: “פאנל ראסופתיות” הכולל את הגנים הידועים ל־Noonan ולמחלות דומות.
- Whole Exome Sequencing (WES) - ריצוף כל האקסום (האזור המקודד). זה בערך 1% מכל ה־DNA, אך בו נמצאות רבות מהמוטציות הגורמות למחלות.
- Whole Genome Sequencing (WGS) - ריצוף הגנום כולו: אקסונים, אינטרונים ואזורים בין־גניים.
כמה וריאנטים מתקבלים?
בריצוף אקסום לאדם מתקבלים בערך 100,000 וריאנטים - כ־100,000 עמדות שבהן הרצף שונה מה־reference. רובם המכריע הם וריאציות נורמליות שמייצגות שונות בין אנשים ואינן גורמות למחלה. האתגר הוא לזהות את השינוי הבודד שגורם למחלה מתוך עשרות אלפי השינויים.
אתגרים
- אזורים חזרתיים - רצפים כמו CAGCAGCAGCAG מקשים על קביעה מדויקת של מספר החזרות, משום שהמקטעים דומים מאוד.
- אזורי GC - אזורים עשירים ב־G וב־C בעייתיים מבחינה כימית ולכן קשים לריצוף.
- Pseudogenes - קושי דומה ל־PCR: מקטע יכול “להתאים” ליותר ממיקום אחד בגנום בשל דמיון גבוה בין רצפים.
חלק ט׳: Variant Classification - הלב של הגנטיקה הקלינית
האתגר המרכזי
זוהה שינוי בגן - השאלה המיידית היא מה המשמעות שלו: האם מדובר בשינוי גורם-מחלה, או בשונות תקינה שלא קשורה למחלה? זאת אחת השאלות המרכזיות בגנטיקה קלינית. בהרצאה הודגש שרוב הגנטיקה עוסקת ב־Variant Classification.
סולם הסיווג (ACMG Guidelines)
| סיווג | משמעות | מה עושים עם זה |
|---|---|---|
| Pathogenic | גורם מחלה (>99% ודאות) | מדווחים, משפיע על טיפול |
| Likely Pathogenic | כנראה גורם מחלה (~90%) | מדווחים, מתייחסים כמו Pathogenic |
| VUS | לא ידוע | מדווחים, אך לא פועלים על סמך זה |
| Likely Benign | כנראה שפיר (~90%) | לרוב לא מדווחים |
| Benign | שפיר (>99%) | לא מדווחים |
VUS - Variant of Unknown Significance
VUS אינו אבחנה. בהרצאה הודגש כי לעיתים קיימת אי־הבנה: עצם הופעת “שינוי גנטי” בדו״ח אינה אומרת שמדובר באבחנה. משמעות VUS היא “לא ידוע” - אין מסקנה האם השינוי גורם למחלה או לא.
למרות זאת, VUS עשוי להיות מדווח משום שייתכן שבעתיד יתווסף מידע: יימצאו אנשים נוספים עם אותו שינוי, או יתפרסם מחקר שיאפשר סיווג מחדש.
כיצד מסווגים? איסוף ראיות
הסיווג מבוסס על גישה של Evidence-Based Medicine: נאספות ראיות בעד ונגד פתוגניות, לכל ראיה ניתן משקל/ניקוד, ולבסוף מתקבלת הכרעה מסכמת.
ראיות חזקות לסיווג וריאנטים
1. שכיחות באוכלוסייה - הראיה החזקה ביותר
כאשר שינוי מופיע בשכיחות גבוהה באוכלוסייה, הוא כנראה לא יכול להיות הסבר למחלה נדירה וקשה. למשל, שינוי שנמצא אצל 50% מהאנשים לא יכול להיות גורם למחלה מונוגנית נדירה - אחרת מחצית מהאוכלוסייה הייתה חולה בה (והיא לא הייתה כל כך נדירה).
כלל אצבע: אם שינוי מופיע ביותר מ־0.5% מהאוכלוסייה, הוא כנראה לא גורם למחלה מונוגנית קשה.
מאגרי מידע לשכיחות:
- gnomAD - המאגר הגדול והמרכזי, כולל מאות אלפי אקסומים וגנומים
- ExAC - גרסה קודמת
- 1000 Genomes - המאגר המקורי
בפועל, ניתן להזין את השינוי ב־gnomAD ולבדוק כמה נשאים קיימים.
2. Loss of Function בגן שפועל ב־Haploinsufficiency
Loss of Function (LoF) הם שינויים שמנטרלים את פעילות הגן, למשל:
- Nonsense - יצירת stop codon מוקדם
- Frameshift - הוספה/מחיקה שמסיטה את מסגרת הקריאה
- מחיקה גדולה - חסר של חלק גדול מן הגן או של הגן כולו
כשהגן פועל במנגנון של Haploinsufficiency (נדרשים שני עותקים תקינים), וריאנט LoF נחשב ראיה חזקה מאוד לפתוגניות.
משקל בסיווג: 8 נקודות - Very Strong evidence.
3. De Novo - הופעה בילד, היעדרות בהורים
שינוי שמופיע בילד חולה ולא קיים בשני הורים בריאים נחשב לראיה חזקה, משום שהוא התרחש ספונטנית ובמקביל הופיעה מחלה.
משקל בסיווג: 4 נקודות - Strong evidence.
חשוב: גם הכיוון ההפוך משמעותי. אם שינוי מורש מהורה בריא, זו ראיה חזקה לשפירות - במיוחד במחלות דומיננטיות קשות.
4. מוטציה שכבר תוארה
כשאותו שינוי בדיוק (או שינוי אחר באותו קודון שמוביל לאותה החלפת חומצת אמינו) כבר תועד בספרות כפתוגני - מדובר בראיה חזקה.
5. עבודות מעבדתיות (Functional Studies)
מחקרים שבודקים בפועל את השפעת השינוי (למשל Western blot, מחקרי RNA ועוד) וכוללים ממצאים כמו חלבון שאינו מיוצר או שאינו מתפקד בצורה תקינה, מספקים ראיה תומכת.
ראיות בינוניות וחלשות
מיקום בחלבון
האם השינוי נמצא באזור חשוב: binding site, דומיין פונקציונלי וכדומה. שינוי באתר קריטי נחשב חשוד יותר.
שימור אבולוציוני
אם חומצת האמינו במיקום זה שמורה לאורך האבולוציה (מדג ועד אדם), קיימת אינדיקציה לחשיבות פונקציונלית ולכן שינוי במיקום כזה עשוי להיות משמעותי יותר.
תוכנות פרדיקציה
כלים כמו SIFT, PolyPhen, CADD מנסים לחזות אם שינוי מזיק על בסיס מבנה החלבון ושימור אבולוציוני.
עם זאת, הודגש בהרצאה: לא להסתמך רק על תוכנות (Softwares) - הן לא תמיד מדויקות, והן מהוות ראיה אחת מתוך מכלול.
תהליך העבודה בפועל
שלב 1: מתקבלים כ־100,000 וריאנטים
לאחר Exome מתקבלת רשימה גדולה במיוחד של וריאנטים.
שלב 2: סינון ראשוני
בוצע סינון על ידי ביואינפורמטיקאים, למשל לפי:
- שכיחות (הסרת וריאנטים שכיחים)
- פנוטיפ (מיקוד בגנים רלוונטיים)
- סוג השינוי (העדפה ל־LoF)
שלב 3: מעבר לרשימה קצרה
בדרך כלל מתקבלים כ־10 וריאנטים שדורשים התייחסות.
שלב 4: סיווג כל וריאנט
נערכת בחינה משותפת של ביואינפורמטיקאים ורופאים, לרוב סביב שאלות כמו:
- מה השכיחות באוכלוסייה?
- האם מדובר ב־De Novo?
- מה סוג השינוי?
- האם קיימת ספרות תומכת?
- על מה מצביעות תוכנות הפרדיקציה?
שלב 5: סיכום וקביעת סיווג
נקבע ניקוד שמוביל לסיווג:
- מעל 10 נקודות ← Pathogenic
- 9-6 נקודות ← Likely Pathogenic
- ביניים ← VUS
כלים שימושיים
- Franklin - כלי חינמי ל־data mining שמציע סיווג על בסיס חיפוש במאגרים
- Varsome - כלי דומה
- ClinVar - מאגר וריאנטים שסווגו בעבר
עם זאת הודגש בהרצאה: בדיקה עצמאית הכרחית - הכלים לא תמיד מדויקים.
חלק י׳: מקרים קליניים
מקרה 1: ילדה עם Split Hand Malformation
הצגה: ילדה שנולדה עם יד וכף רגל לא תקינות - “יד שסועה” (Split Hand). מדובר במום מולד מסוים.
הבדיקה: Trio Exome (הילדה ושני ההורים).
הממצא: וריאנט בגן TP63 - גן המוכר כקשור ל־Split Hand.
תהליך הסיווג:
- De Novo (לא מורש מההורים) ✓
- ממוקם באזור שבו מוכרות מוטציות פתוגניות רבות ✓
- נדיר באוכלוסייה ✓
- תוכנות פרדיקציה מצביעות על פגיעה ✓
- תועדו משפחות נוספות עם אותו שינוי ואותו פנוטיפ ✓
מסקנה: Pathogenic - זו ההבחנה.
מקרה 2: ילד עם איחור התפתחותי - Exome תקין
הצגה: ילד בן 4 עם איחור התפתחותי; ההורים בריאים.
הבדיקה: Trio Exome.
הממצא: לא נמצא ממצא מסביר.
מה המשמעות? אין פירוש הדבר שאין בעיה גנטית. המשמעות היא שלא נמצא שינוי מתאים במסגרת הבדיקה. ייתכן למשל:
- מוטציה באזור שלא כוסה
- מוטציה בגן חדש שעדיין אינו מוכר
- ייתכן גם שמדובר בגורם שאינו גנטי (אך אין ודאות)
מקרה 3: ילד עם VUS
הצגה: ילד בן 4 עם איחור התפתחותי ואוטיזם.
הבדיקה: Exome.
הממצא: וריאנט בגן SHANK3 - גן שקשור לאוטיזם ולאיחור התפתחותי.
הבעיה: לאחר סיווג מתקבל VUS - אין די ראיות לקביעה פתוגניות.
מה עושים? הממצא מדווח, תוך הסבר ברור למשפחה שמדובר בממצא שאינו אבחנה, וייתכן שבעתיד יצטבר מידע שיאפשר סיווג מחדש.
נקודה שהודגשה בהרצאה: קיימת נטייה לפרש “שינוי בגן שקשור לפנוטיפ” כאבחנה, אך זו Selection Bias - וריאנטים שעולים לדיון הם מלכתחילה כאלה שנמצאים בגנים שנבחרו בגלל הפנוטיפ. עצם ההתאמה לקטגוריית הגנים לא מוכיחה שהווריאנט הוא גורם המחלה.
חלק י״א: Secondary Findings ו־Incidental Findings
Secondary Findings - ממצאים משניים
מה זה?
מדובר ברשימה של 84 גנים שנבדקים באופן אקטיבי בכל בדיקת Exome או Genome, גם כאשר אינם קשורים לשאלה הקלינית המקורית.
למה עושים את זה?
משום שמדובר בגנים Actionable - כלומר, אם מתגלה בהם מוטציה, קיימת משמעות מעשית ויש מה לעשות מבחינת מניעה/מעקב/טיפול.
דוגמאות:
- BRCA1/2 - סרטן שד ושחלות: מעקב הדוק ואפשרות לניתוח מניעתי.
- גנים של Lynch syndrome - סרטן מעי: קולונוסקופיות בתדירות גבוהה.
- גנים לאריתמיות - סיכון למוות לבבי פתאומי: אפשרויות טיפול מניעתי.
כללים חשובים
- הסכמה מראש: נשאלים מראש האם קיים רצון לקבל מידע על Secondary Findings.
- דיווח בילדים: לא מדווחים בילדים ממצאים של מחלות “מבוגרים בלבד”. לדוגמה, אם מתגלה BRCA בילדה בת 5 - לא מדווחים. הנימוק: זכות לאוטונומיה; ההחלטה אם לקבל מידע כזה אמורה להתקבל בגיל 18, ולא על ידי ההורים.
חריג: כאשר הממצא רלוונטי לגיל הילדות (למשל APC שיכול לגרום לפוליפים כבר סביב גיל 12) - כן מדווחים.
דוגמה: APC ושתי מוטציות שונות
- מוטציה קלאסית ב־APC - גורמת ל־Familial Adenomatous Polyposis. לפי הדוגמה, 100% מהנשאים יפתחו סרטן מעי, ומתחילים מעקב מגיל 8. זה מוגדר Actionable ולכן מדווחים.
- מוטציה I1307K ב־APC - מעלה סיכון פי 2 בלבד (מ־2% ל־4%). לפי הדוגמה, אין שינוי מעשי במעקב (ממילא קיימת המלצה לכלל האוכלוסייה לקולונוסקופיה מגיל 50), ולכן זה מוגדר לא Actionable ולא מדווחים.
Incidental Findings - ממצאים מקריים
אלו ממצאים שעולים במקרה, ללא חיפוש יזום.
דוגמה: נבדק גן הקשור לפרכוסים, ובמהלך הניתוח מזוהה במקרה מוטציה בגן הקשור לחירשות - ממצא שאינו קשור לשאלה המקורית אך התגלה.
גם כאן, לפי ההרצאה, הנוהל הוא לשאול מראש האם קיים רצון לקבל מידע על ממצאים מסוג זה.
חלק י״ב: סוגיות אתיות
מקרה לדוגמה: BRCA ב־Panel ממוקד
המקרה: ילדה קטנה מקהילה חרדית עם חשד ל־Noonan syndrome. הוזמן Panel ממוקד שכולל רק גנים של Noonan, במטרה להימנע מחשיפה למידע נוסף שעלול להפריע לשידוכים.
מה קרה בפועל: המעבדה עדכנה שבמהלך העבודה “בטעות” נצפה שלילדה יש BRCA, אך הוסיפה: “זה לא יופיע בדו״ח”.
הדילמה:
- המידע לא התבקש על ידי הילדה או הוריה
- חשיפתו תפגע באוטונומיה שלהם
- מנגד, מדובר במידע שעשוי להציל חיים בעתיד
הפעולה שנעשתה: האם הוזמנה והומלץ לה לבצע בדיקת BRCA משום שהיא אשכנזייה (כהמלצה כללית לנשים אשכנזיות). לאחר חתימה על טופס, היא סירבה להיבדק.
התוצאה: הצוות הרפואי יודע שלילדה יש BRCA אבל הוא לא יכול לעשות דבר על בסיס המידע. ייתכן שבעתיד הילדה תבחר להיבדק, אך כרגע אין אפשרות ליידע אותה או את משפחתה.
העיקרון: אוטונומיה
גם לילדים קיימות זכויות, ובפרט הזכות להחליט על עתידם הגנטי כאשר יגיעו לגיל שבו ניתן להבין את המשמעות. כמו שלא ניתן לקחת ילד מהרחוב ולבדוק אותו ל־BRCA, כך גם לא אמור להתאפשר “להשיג” מידע גנטי על ילד דרך בדיקה אחרת באופן עקיף.
חלק י״ג: Rapid Whole Genome - BabySeq
מדובר בריצוף גנום מהיר לתינוקות בטיפול נמרץ, כאשר התשובה מתקבלת בתוך 5 ימים במקום שבועות.
למי זה מיועד?
לתינוק שנולד בזמן ונראה תקין, אך לאחר מכן חלה הידרדרות פתאומית - פרכוסים, כשל נשימתי, בעיות מטבוליות - ללא הסבר ברור.
למה Genome ולא Exome?
- מהיר יותר - מדלג על שלב ה־Capture
- כיסוי טוב יותר של האקסום
- מאפשר לזהות גם ממצאים מחוץ לאקסום
מקרה חיובי: הצלחה
- הצגה: תינוק בן 48 שעות עם פרכוסים רציפים.
- ממצא: מוטציה בגן הקשור ל־B6-Dependent Epilepsy - מצב שבו הפרכוסים נפסקים לאחר מתן ויטמין B6.
- תוצאה: ניתן ויטמין B6, והתינוק חזר להיות תקין - חיים ניצלו.
מקרה שלילי: גם זה עוזר
הצגה: תינוק עם פרכוסים קשים בדומה למקרה הקודם. ממצא: מוטציה בגן הקשור ל־Epileptic Encephalopathy - מחלה עם פרכוסים עמידים לטיפול, ללא אפשרות טיפולית יעילה. תוצאה: הופסק ניסיון בטיפולים אגרסיביים לאחר שהובהר שלא צפויה תועלת. הדבר סייע להורים להתמודד ולקבל תחושת סגירת מעגל.
המסר
גם כאשר אין טיפול, לאבחנה יש ערך. היא עשויה לסייע ב־
- הפסקת בירור מיותר וכואב
- מתן תשובות להורים
- תכנון הריונות עתידיים
- קבלת החלטות לגבי המשך או הפסקת טיפול
חלק י״ד: מושגים חשובים נוספים
Locus Heterogeneity
מצב שבו מוטציות בגנים שונים גורמות לאותה מחלה.
דוגמה קלאסית: Noonan syndrome Noonan יכול להיגרם ממוטציות בגנים שונים, למשל:
- PTPN11
- SOS1
- RAF1
- KRAS
- ועוד גנים נוספים
ההסבר הוא שכל הגנים הללו משתתפים באותו מסלול ביולוגי - RAS-MAPK pathway. פגיעה בנקודות שונות באותו מסלול יכולה להוביל לתמונה קלינית דומה.
RASopathies
קבוצת מחלות הנגרמות ממוטציות במסלול RAS-MAPK, ובהן:
- Noonan syndrome
- Costello syndrome
- CFC (Cardio-Facio-Cutaneous)
- Neurofibromatosis type 1
למחלות האלה יש מאפיינים חופפים, כמו קומה נמוכה, מומי לב, ודיסמורפיזם פנים.
פליאוטרופיה (Pleiotropy) - מוטציה בגן אחד (NF1) גורמת לביטויים קליניים מרובים.
Noonan syndrome - “ה־Turner האוטוזומלי”
מאפיינים אופייניים:
- קומה נמוכה
- צוואר רחב
- אוזניים נמוכות ומסובבות
- מום לבבי
ההבדל לעומת Turner:
- Turner - 45,X (חסר כרומוזום X)
- Noonan - תורשה אוטוזומלית (מוטציה בגן)
- Turner - מום באאורטה, למשל Coarctation
- Noonan - מום במוצא הריאה, למשל Pulmonic stenosis
סיכום נקודות למבחן
מה חשוב לזכור
- שכיחות באוכלוסייה היא הראיה החזקה ביותר לסיווג וריאנטים: וריאנט שכיח אינו מתאים כמסביר מחלה נדירה.
- De Novo במחלה דומיננטית נחשב ראיה חזקה מאוד לפתוגניות.
- Loss of Function בגן Haploinsufficient הוא ממצא חזק מאוד (8 נקודות).
- VUS אינו אבחנה - המשמעות היא “לא ידוע”.
- Coverage = האם האזור רוצף; Depth = כמה פעמים כל עמדה נקראה.
- Exome ≈ 1% מהגנום (האזור המקודד בלבד); Genome כולל את כל הרצף.
- Secondary Findings = רשימת 84 גנים Actionable שנבדקים באופן יזום.
- BRCA בילדים לא מדווח מטעמי אוטונומיה, כדי לאפשר החלטה עצמאית בגיל 18.
- הבדלים בין שיטות:
- Western blot = חלבונים;
- Northern = RNA;
- Southern = DNA.
- Sanger מוגבל לכ־~500 bp, בעוד NGS מרצף מיליוני מקטעים במקביל.
מה יכול להופיע בשאלה
- וריאנט עם נתונים (שכיחות, האם De Novo, סוג שינוי) ושאלה האם הסיווג מתאים ל־Pathogenic
- השוואה בין סוגי בדיקות (Panel / Exome / Genome, או Sanger מול NGS)
- שאלות על דיווח: מתי מדווחים ומתי לא
- מושגים מרכזיים: Coverage, Depth, VUS
נאמר בהרצאה שזה לא יהיה מסובך: המידע הדרוש צפוי להינתן בשאלה, והדגש הוא על הבנת העקרונות. מניסיון, עדיף לשנן כמה שיותר, במיוחד פרטים קטנים וטכניים.
שאלות תרגול מג׳ונרטות
שאלות תרגול בגנטיקה על בסיס תרגול 8: PCR, אנזימי רסטריקציה, RFLP, ג׳ל אלקטרופורזה, ריצוף סנגר, Blotting, NGS, pipeline ביואינפורמטי, סיווג ACMG, Actionable Findings.
שאלה 1: PCR - שלבי הריאקציה
מה הסדר הנכון של שלושת השלבים במחזור PCR?
- Annealing → Denaturation → Extension
- Denaturation → Annealing → Extension
- Extension → Denaturation → Annealing
- Denaturation → Extension → Annealing
פתרון
התשובה הנכונה היא (2).
כל מחזור PCR כולל שלושה שלבים:
- Denaturation (~95°C) - חימום ה־DNA כדי להפריד בין שני הגדילים
- Annealing (~55-65°C) - חיבור ה־Primers לרצפים הקומפלמנטריים על הגדיל
- Extension (~72°C) - Taq Polymerase מאריך את הגדיל החדש בכיוון 5’←3’
מבצעים 30-20 מחזורים ← מיליוני עותקים של המקטע הרצוי.
למה האחרות שגויות:
- (1) לא ניתן לחבר Primers לפני שהגדילים נפרדו.
- (3) ו־(4) - Extension חייב להיות אחרון, אחרי שה־Primers כבר מחוברים.
| מקור: תרגול 8 – PCR | שיעור 10 – שיטות אבחון גנטי |
שאלה 2: PCR - רכיבים
חוקר רוצה לבצע PCR אך שכח להוסיף Primers. מה יקרה?
- ה־DNA ישוכפל כרגיל אך לאט יותר
- Taq Polymerase ישכפל את כל הגנום
- לא יתרחש שכפול כלל
- רק הגדיל המוביל (Leading Strand) ישוכפל
פתרון
התשובה הנכונה היא (3).
DNA Polymerase (כולל Taq) לא יכול להתחיל סינתזה מאפס - הוא רק מאריך גדיל קיים. לכן חובה שיהיה Primer (רצף קצר של DNA/RNA) שנקשר לגדיל התבנית ומספק קצה 3’-OH חופשי שממנו האנזים יכול להתחיל.
- Taq Polymerase זקוק ל־Primer כדי להתחיל
ללא Primers ← אין התחלה ← אין תוצר PCR.
הערה חשובה מהתרגול: ה־Primers צריכים להיות ספציפיים וייחודיים. אם הם קצרים מדי - יתחברו למקומות רבים בגנום. צריך שני Primers בכיוונים מנוגדים (Forward ו־Reverse).
| מקור: תרגול 8 – PCR | שיעור 10 – PCR |
שאלה 3: ג׳ל אלקטרופורזה - עקרון הפעולה
בג׳ל אלקטרופורזה, לאן ינדוד מקטע DNA גדול יותר?
- רחוק יותר מהמקור - מקטעים גדולים נודדים מהר יותר
- קרוב יותר למקור - מקטעים גדולים נודדים לאט יותר
- DNA לא נודד בג׳ל - צריך Probe ספציפי
- קרוב יותר למקור - DNA גדול יותר טעון יותר שלילי ולכן נמשך פחות
פתרון
התשובה הנכונה היא (2).
ג׳ל אלקטרופורזה מפריד מקטעי DNA לפי גודל:
- DNA טעון שלילית (בגלל קבוצות הפוספט) ← נודד לכיוון הקוטב החיובי
- הג׳ל פועל כמסננת - מקטעים קטנים עוברים בקלות ונודדים רחוק
- מקטעים גדולים מתקשים לעבור ונשארים קרוב למקור
שימוש: לאחר PCR, מריצים את התוצרים על ג׳ל כדי לראות אם ההגברה הצליחה, ומה גודל המקטעים.
שאלה 4: אנזימי רסטריקציה - מהות
מה מאפיין אנזים רסטריקציה?
- אנזים שמבודד חלבונים ברצפים ספציפיים
- אנזים שמבודד RNA ברצפים ספציפיים
- חלבון מבקטריות שחותך DNA ברצפים ספציפיים
- אנזים שמחבר מקטעי DNA - ההיפך מ־Ligase
פתרון
התשובה הנכונה היא (3).
ויוצר מקטעים עם קצוות ידועים.
Restriction Enzyme (אנדונוקליאז רסטריקציה):
- חלבון מבקטריות שמזהה רצף ספציפי ב־DNA וחותך אותו
- מייצר מקטעי DNA עם קצוות ידועים
- דוגמאות מהתרגול:
| אנזים | אתר חיתוך |
|---|---|
| EcoRI | G↓AATTC |
| BamHI | G↓GATCC |
| HaeIII | GG↓CC |
| HhaI | GCG↓C |
| HindIII | A↓AGCTT |
שימוש בגנטיקה: זיהוי מוטציות - אם מוטציה משנה את אתר החיתוך, האנזים לא יחתוך ← מקטעים בגודל שונה בג׳ל.
| מקור: תרגול 8 – Restriction Enzymes | שיעור 10 – אנזימי רסטריקציה |
שאלה 5: RFLP - זיהוי מוטציה
ברצף Wild Type יש אתר חיתוך של HaeIII (GGCC). מוטציה C>A משנה את הרצף ל־GGCA. מה יקרה כשנחתוך עם HaeIII?
- שני הרצפים ייחתכו באותו אופן
- ה־Wild Type ייחתך, המוטנטי לא
- המוטנטי ייחתך, ה־Wild Type לא
- לא ניתן להשתמש ב־HaeIII לזיהוי מוטציה זו
פתרון
התשובה הנכונה היא (2).
עקרון RFLP (Restriction Fragment Length Polymorphism):
- HaeIII מזהה וחותך את הרצף GGCC בלבד
- Wild Type (GGCC) ← ייחתך ← שני מקטעים קצרים
- מוטנטי (GGCA) ← לא ייחתך ← מקטע אחד ארוך
מה רואים בג׳ל:
- WT: שני פסים (מקטעים קטנים, נודדים רחוק)
- מוטנטי: פס אחד (מקטע גדול, נודד קרוב)
- הטרוזיגוט: שלושה פסים (אלל אחד נחתך, אחד לא)
זהו בדיוק העיקרון שהוצג בתרגול עם הדוגמה של GGCC ← C>A.
מקור: תרגול 8 – PCR and RFLP
שאלה 6: RFLP - פרשנות תוצאות ג׳ל במשפחה
במשפחה עם מחלה אוטוזומלית רצסיבית, בוצעה בדיקת RFLP. הילד החולה מציג פס אחד בלבד בג׳ל. שני ההורים מציגים שלושה פסים. מה גנוטיפ ההורים?
- שניהם הומוזיגוטים לאלל התקין (+/+)
- שניהם הטרוזיגוטים (+/-)
- שניהם הומוזיגוטים לאלל המוטנטי (-/-)
- האב +/+ והאם -/-
פתרון
התשובה הנכונה היא (2).
פרשנות תוצאות RFLP בג׳ל:
| גנוטיפ | מספר פסים | הסבר |
|---|---|---|
| +/+ (הומוזיגוט תקין) | 2 פסים | שני האללים נחתכים |
| +/- (הטרוזיגוט/נשא) | 3 פסים | אלל אחד נחתך (2 מקטעים) + אלל אחד לא (1 מקטע) |
| -/- (הומוזיגוט מוטנטי) | 1 פס | אף אלל לא נחתך |
- הילד חולה (-/-) ← 1 פס
- ההורים נשאים (+/-) ← 3 פסים
- זה מתאים בדיוק לתורשה אוטוזומלית רצסיבית
מקור: תרגול 8 – RFLP בעץ משפחה
שאלה 7: זיהוי נשאים של CF בעזרת חיתוך אנזימטי
בבדיקת נשאות ל־CFTR, מה משמעות ה־UC (Uncut) ביחס ל־NTC?
- UC = תוצר PCR שלא עבר חיתוך אנזימטי - משמש כבקרה לגודל המקטע המלא
- UC = תוצאת חיתוך חלקית - משמשת לזיהוי הטרוזיגוטים
- UC ו־NTC הם אותו דבר - משמשים לבקרת זיהום
- UC = מקטע שנחתך לחלוטין - משמש לזיהוי הומוזיגוטים
פתרון
התשובה הנכונה היא (1).
בבדיקת RFLP משתמשים בבקרות:
| קיצור | משמעות | תפקיד |
|---|---|---|
| UC (Uncut) | תוצר PCR ללא חיתוך אנזימטי | מראה את גודל המקטע המלא - בקרה |
| NTC (No Template Control) | PCR ללא DNA תבנית | מוודא שאין זיהום בריאקציה |
| M (Marker) | סולם גדלים | מאפשר לקבוע גודל מקטעים |
בדוגמה מהתרגול (CFTR):
- הראו חיתוך אנזימטי לשינויים שונים בגן CFTR
- c.3718-2477C>T ו־c.3266G>A (p.Trp1089Ter, W1089X)
- ניתן להבחין בין נשאים (הטרוזיגוטים - פסים של חתוך + לא חתוך) לבריאים (רק פסים של חתוך או רק לא חתוך)
שאלה 8: ריצוף סנגר - עקרון
מה תפקידם של ה־ddNTPs (Dideoxynucleotides) בריצוף סנגר?
- מאיצים את הסינתזה של DNA Polymerase
- גורמים לסיום מוקדם של הסינתזה במקום ספציפי
- מחליפים לחלוטין את ה־dNTPs הרגילים
- משמשים כ־Primers לריאקציה
פתרון
התשובה הנכונה היא (2).
- כל ddNTP עוצר את השרשרת כי חסר את קבוצת ה־OH על הפחמן 3’ ← אין אפשרות להוסיף נוקלאוטיד נוסף
עקרון ריצוף סנגר (Dideoxy Sequencing):
- ddNTPs חסרים את קבוצת ה־OH על הפחמן 3’ ← אין אפשרות להוסיף נוקלאוטיד נוסף
- כאשר ddNTP משתלב באקראי במקום dNTP ← השרשרת נעצרת
- נוצרים מקטעים בכל אורך אפשרי, כל אחד מסתיים ב־ddNTP ספציפי
בג׳ל: המקטעים נפרדים לפי גודל ← קוראים את הרצף מלמטה למעלה (מהקצר לארוך) = הרצף בכיוון 5’←3’
בתרגול הוצגה דוגמה לקריאת ג׳ל סנגר:
3’- AGATTTCCAGGTCCCCAGTGGAAAGCTT -5’
5’- TCTAAAGGTCCAGGGGTCACCTTTCGAA -3’
| מקור: תרגול 8 – Sanger Sequencing | שיעור 10 – ריצוף DNA |
שאלה 9: קריאת כרומטוגרם - Hemizygote מול Heterozygote
בריצוף סנגר של גן על כרומוזום X, מה ההבדל בין כרומטוגרם של גבר חולה (Hemizygote) לאישה נשאית (Heterozygote)?
- אין הבדל - שניהם מראים שני שיאים (peaks) באותה עמדה
- ה־Hemizygote מראה שיא אחד של האלל המוטנטי בלבד, ה־Heterozygote מראה שני שיאים חופפים באותה עמדה
- ה־Heterozygote מראה שיא אחד בלבד
- הכרומטוגרם לא יכול להבחין בין השניים
פתרון
התשובה הנכונה היא (2).
בדוגמה מהתרגול (SED - Spondyloepiphyseal Dysplasia Tarda):
גן SEDL על כרומוזום X, מוטציה c.346G>T (p.E116X):
- בן דוד חולה (Hemizygote - XᵃY): שיא אחד בלבד (T) - יש לו רק עותק אחד של X
- אם הנבדקת (Heterozygote - XᴬXᵃ): שני שיאים חופפים באותה עמדה - G (אלל תקין) + T (אלל מוטנטי)
- אחות הנבדקת (Heterozygote): אותו דפוס - שני שיאים
כלל: Hemizygous = שיא נקי אחד. Heterozygous = double peak.
מקור: תרגול 8 – ריצוף גן SEDL
שאלה 10: זיהוי מוטציית חסר (Deletion) בכרומטוגרם
בריצוף של חולה עם PKD6 נמצאה מוטציה c.720_721del. איך תיראה תוצאת הריצוף ב־Heterozygote?
- שני שיאים חופפים בנקודה אחת בלבד
- כרומטוגרם נקי לחלוטין ללא שינוי
- מנקודת ה־Deletion ואילך - הכרומטוגרם הופך לשני רצפים חופפים (מעורבב) כי מסגרות הקריאה כבר לא מסונכרנות
- המוטציה לא ניתנת לזיהוי בריצוף סנגר
פתרון
התשובה הנכונה היא (3).
בדוגמת PKD6 מהתרגול (גן DNAJB11, c.720_721del):
ב־Heterozygote:
- לפני נקודת ה־Deletion - שני האללים מסונכרנים ← כרומטוגרם נקי
- מנקודת ה־Deletion ואילך - אלל אחד קצר ב־2 בסיסים ← מסגרות הקריאה מוסטות ← שני רצפים שונים נקראים בו־זמנית ← כרומטוגרם מעורבב (double peaks בכל עמדה)
השוואה ל־Wild Type:
- WT מראה כרומטוגרם נקי לכל האורך - שיא אחד בכל עמדה
זה הסימן האופייני ל־Indel (Insertion/Deletion) בהטרוזיגוט בריצוף סנגר.
שאלה 11: שיטות Blotting - התאמה
חוקרים רוצים לבדוק רמות ביטוי של mRNA בעכבר מוטנטי. באיזו שיטה ישתמשו?
- Southern Blot
- Northern Blot
- Western Blot
- PCR רגיל
פתרון
התשובה הנכונה היא (2).
| שיטה | בודקת | Probe |
|---|---|---|
| Southern Blot | DNA | DNA או RNA fragment |
| Northern Blot | RNA (mRNA) | DNA או RNA fragment |
| Western Blot | חלבון (Protein) | Antibody (נוגדן) |
טריק לזכירה: S-N-W ← D-R-P (סדר אלפביתי של השיטות תואם לסדר של DNA-RNA-Protein)
שיטות נוספות:
- PCR - מגביר DNA (לא מודד רמות ביטוי של mRNA)
- RT-PCR (Reverse Transcription PCR) - ממיר RNA ל־cDNA ואז מגביר ← גם יכול לבדוק ביטוי mRNA
מהתרגול: חוקרים שרוצים להשוות ביטוי mRNA = Northern Blot. ביטוי חלבון = Western Blot.
| מקור: תרגול 8 – Blotting | שיעור 10 – שיטות Blotting |
שאלה 12: Western Blot מול Northern Blot
מדוע תיתכן תוצאה שבה Northern Blot מראה ביטוי תקין של mRNA אך Western Blot מראה חלבון חסר?
- זה בלתי אפשרי - אם ה־mRNA תקין, החלבון חייב להיות תקין
- ייתכן שהמוטציה פוגעת בתרגום או ביציבות החלבון אך לא בשעתוק
- Western Blot תמיד מראה את אותן תוצאות כמו Northern
- הסיבה היחידה היא שגיאת מעבדה
פתרון
התשובה הנכונה היא (2).
mRNA תקין + חלבון חסר יכול לנבוע מ:
- מוטציית Nonsense (Stop Codon מוקדם) ← mRNA נוצר אך חלבון מקוצר ומפורק (NMD - Nonsense Mediated Decay)
- בעיה בPost-translational modification ← חלבון לא יציב ומתפרק
- מוטציה באזור 5’UTR או Kozak ← בעיה באתחול התרגום
חשוב: השעתוק (Transcription) והתרגום (Translation) הם שלבים נפרדים. פגיעה בתרגום לא בהכרח פוגעת ב־mRNA.
שאלה 13: Shotgun Sequencing
מהו העיקרון של Shotgun Sequencing?
- ריצוף הגנום מקצה אחד לשני ברצף, ריצוף כל מקטע עד להשלמת הגנום, ואז הרכבה לפי סדר הכרומוזומים
- חיתוך הגנום למקטעים אקראיים קטנים, ריצוף כל מקטע, והרכבה מחדש לפי חפיפות ברצפים
- ריצוף רק של האקסונים (Exome), חיתוך רק האזורים המקודדים, ואז הרכבה לפי סדר הכרומוזומים
- שימוש ב־Restriction Enzymes לחיתוך, ריצוף כל מקטע, והרכבה מחדש לפי חפיפות ברצפים
פתרון
התשובה הנכונה היא (2).
Shotgun Sequencing (מהתרגול, Figure 14-4):
- חיתוך - עותקים רבים של הגנום נחתכים למקטעים אקראיים קטנים
- ריצוף - כל מקטע מרוצף בנפרד
- חפיפה (Overlap) - מחפשים רצפים זהים בין מקטעים שונים
- הרכבת Contigs - מקטעים חופפים מורכבים ליחידות רציפות
- רצף קונצנזוס - מיזוג ה־Contigs לרצף מלא של כל כרומוזום
למה האחרות שגויות:
- (1) Chromosome Walking = שיטה אחרת, איטית יותר.
- (3) Shotgun מרצף את כל הגנום, לא רק Exome.
- (4) החיתוך הוא אקראי (מכני/אנזימטי), לא עם Restriction Enzymes ספציפיים.
שאלה 14: NGS - Next Generation Sequencing
מה היתרון המרכזי של שיטת Illumina על ריצוף סנגר?
- Illumina מדויק יותר לכל קריאה בודדת
- Illumina מרצף מיליוני מקטעים במקביל
- Illumina לא דורש Primers כלל
- Illumina זול יותר לריצוף גן בודד
פתרון
התשובה הנכונה היא (2).
NGS (Illumina) - שלושה שלבים עיקריים (Figure 14-6):
- Library Construction - הכנת ספרייה של מקטעי DNA עם Adapters
- Cluster Formation - קשירת מקטעים ל־Flow Cell והגברה ל־Clusters
- Sequencing by Synthesis - ריצוף מיליוני Clusters בו־זמנית
(Massively Parallel Sequencing)
למה האחרות שגויות:
- (1) לקריאה בודדת, סנגר מדויק יותר. NGS מפצה ב־Coverage גבוה.
- (3) NGS כן דורש Adapters/Primers.
- (4) לגן בודד, סנגר עדיין זול ומהיר יותר. NGS משתלם ל־Exome/Genome.
מתי נעדיף NGS?
- פאנלים של עשרות-מאות גנים
- Whole Exome Sequencing (WES)
- Whole Genome Sequencing (WGS)
| מקור: תרגול 8 – NGS | שיעור 10 – שיטות ריצוף |
שאלה 15: Pipeline ביואינפורמטי - סדר השלבים
מה הסדר הנכון של שלבי ניתוח NGS?
- Variant Calling → Base Calling → Alignment → Filtering
- Base Calling → Alignment → Variant Calling → Filtering/Annotation → Causal Variant
- Alignment → Base Calling → Filtering → Variant Calling
- Filtering → Variant Calling → Alignment → Base Calling
פתרון
התשובה הנכונה היא (2).
Pipeline ביואינפורמטי (מהתרגול, Figure 1):
| שלב | תהליך | תוצאה |
|---|---|---|
| 1. Base Calling | המרת אותות ריצוף לנוקלאוטידים | קובץ FASTQ (קריאות קצרות) |
| 2. Alignment | יישור הקריאות לגנום הייחוס | קובץ SAM/BAM |
| 3. Variant Calling | זיהוי הבדלים מגנום הייחוס | קובץ VCF (רשימת וריאנטים) |
| 4. Filtering/Annotation | סינון לפי מאגרי מידע | רשימה מצומצמת של וריאנטים מתועדים |
| 5. Causal Variant | זיהוי הווריאנט הגורם למחלה | וריאנט סיבתי |
שאלה 16: מושגים בסיסיים - Wild Type מול Variant
מה ההבדל בין Wild Type, Variant, ו־Pathogenic Variant?
- כולם מילים נרדפות לאותו דבר
- Wild Type = האלל הנפוץ, Variant = אלל שונה (ללא סיווג קליני), Pathogenic Variant = שינוי גנטי גורם מחלה
- Wild Type = מוטציה, Variant = תקין, Pathogenic Variant = שינוי גנטי גורם מחלה
- Wild Type = האלל הנפוץ, Variant = אלל שונה (ללא סיווג קליני), Pathogenic Variant = כל SNP
פתרון
התשובה הנכונה היא (2).
הגדרות מהתרגול:
| מונח | הגדרה |
|---|---|
| Locus | אזור ספציפי בכרומוזום - גן שלם או נוקלאוטיד בודד |
| Allele | וריאציות של רצף ה־DNA בלוקוס מסוים |
| Wild Type | האלל הנפוץ באוכלוסייה |
| Variant | אלל שונה מ־Wild Type - ללא התייחסות לסיווג קליני |
| Pathogenic Variant | שינוי גנטי גורם מחלה |
| Mutation | שינוי ברצף DNA - ללא התייחסות לסיווג קליני |
| SNP | שינוי בנוקלאוטיד בודד - ללא התייחסות לסיווג קליני |
חשוב: SNP ≠ פתוגני בהכרח. וריאנט ≠ מחלה בהכרח. רק Pathogenic Variant = גורם מחלה.
מקור: תרגול 8 – מושגים
שאלה 17: סיווג ACMG - חמש הקטגוריות
לפי כללי ACMG, מה המשמעות של “Likely Pathogenic”?
- הווריאנט בוודאות גורם מחלה
- סיכוי של מעל 90% שהווריאנט גורם מחלה
- לא ידוע אם הווריאנט גורם מחלה - צריך לחכות
- הווריאנט בוודאות לא גורם מחלה
פתרון
התשובה הנכונה היא (2).
5 קטגוריות ACMG (מהתרגול):
| קטגוריה | משמעות |
|---|---|
| Pathogenic | גורם מחלה |
| Likely Pathogenic | >90% סיכוי לגרום מחלה |
| VUS (Variant of Unknown Significance) | לא ידוע |
| Likely Benign | >90% סיכוי שלא גורם מחלה |
| Benign | שפיר |
חשוב קלינית:
- Pathogenic / Likely Pathogenic ← ניתן לפעול על בסיסם (שינוי ניהול קליני)
- VUS ← לא משנים ניהול קליני, עוקבים ומחכים למידע נוסף
- Likely Benign / Benign ← אין משמעות קלינית
| מקור: תרגול 8 – Variant Calling, ACMG | שיעור 10 – ACMG |
שאלה 18: קריטריונים ACMG - Very Strong Evidence
מהו הקריטריון PVS1?
- מוטציה De Novo שאושרה
- מחקר פונקציונלי שמוכיח השפעה מזיקה
- Null Variant בגן שבו LOF הוא מנגנון ידוע של מחלה
- הווריאנט נדיר ולא מופיע ב־gnomAD
פתרון
התשובה הנכונה היא (3).
PVS1 (Pathogenic Very Strong 1) - הקריטריון החזק ביותר לפתוגניות:
Null Variant = מוטציה שגורמת לאובדן מוחלט של תפקוד הגן:
- Nonsense (Stop Codon מוקדם)
- Frameshift (הזזת מסגרת קריאה)
- Canonical splice (±1 or ±2)
- Initiation codon (פגיעה בקודון ההתחלה)
- Multi-exon deletion
אזהרות (Caveats) מהתרגול:
- לא תקף אם LOF אינו מנגנון ידוע של מחלה באותו גן
- זהירות עם מוטציות בקצה ה־3’ ביותר של הגן
- זהירות עם מוטציות Splice שגורמות ל־Exon Skipping אך משאירות את שאר החלבון שלם
למה האחרות שגויות:
- (1) De Novo = PS2 (Strong Evidence)
- (2) מחקר פונקציונלי = PS3 (Strong Evidence)
- (4) נדיר ב־gnomAD = PM2 (Moderate Evidence)
שאלה 19: מקרה קליני - סיווג ACMG
בת 28 עם כליות פוליציסטיות, לאבא קליניקה דומה. נמצא שינוי בגן PKD1: c.12683G>C p.Arg4228Pro. השינוי נדיר, שמור אבולוציונית, תוכנות פרדיקציה חוזות השפעה מזיקה, ומחקר פונקציונלי הוכיח שינוי משמעותי במבנה החלבון. השינוי נמצא גם אצל האב. מה הסיווג הצפוי?
- VUS - אין מספיק ראיות
- Likely Benign - השינוי לא גורם נזק
- Pathogenic - עם PS3, PM2, PP1, PP3
- Benign - כי נמצא גם אצל האב
פתרון
התשובה הנכונה היא (3).
ניתוח לפי קריטריוני ACMG:
| קריטריון | עוצמה | הסבר |
|---|---|---|
| PS3 | Strong | מחקר פונקציונלי הראה שינוי משמעותי במבנה החלבון |
| PM2 | Moderate | נדיר - לא מופיע ב־gnomAD |
| PP1 | Supporting | Co-segregation - נמצא גם אצל האב שחולה |
| PP3 | Supporting | תוכנות פרדיקציה (PolyPhen-2, SIFT, MutationTaster) חוזות השפעה מזיקה |
סיכום: Strong + Moderate + 2 Supporting ← מספיק ל־Pathogenic.
למה האחרות שגויות:
- (1) יש מספיק ראיות - לא VUS.
- (2) הראיות תומכות בפתוגניות, לא שפירות.
- (4) שהשינוי נמצא גם אצל האב שחולה = PP1 (Co-segregation) - ראיה בעד פתוגניות, לא נגד!
שאלה 20: Actionable (Secondary) Findings
בריצוף אקסום של אישה עם הפרעת קצב נמצאו:
- שינוי פתוגני ב־BRCA1
- VUS ב־BRCA2
- שינוי פתוגני לליקוי שמיעה בגיל מבוגר
- שינוי בגן לאלצהיימר בגיל צעיר
על מה חובה לדווח?
- על כל ארבעת השינויים כי כולם פתוגניים
- רק על BRCA1 - כי יש פעולות מניעה/אבחון מוקדם שניתן לבצע
- רק על VUS ב־BRCA2 כי יכול להיות פתוגני
- על BRCA1 ועל אלצהיימר כי שניהם פתוגניים
פתרון
התשובה הנכונה היא (2).
Actionable (Secondary) Findings - ממצאים שלא קשורים לסיבת ההפניה אך יש להם משמעות קלינית שניתן לפעול על בסיסה:
| ממצא | לדווח? | סיבה |
|---|---|---|
| BRCA1 פתוגני | כן ✓ | ניתן לבצע מעקב מוגבר, MRI, ניתוח מניעתי |
| VUS ב־BRCA2 | לא ✗ | VUS = לא משנה ניהול קליני |
| ליקוי שמיעה בגיל מבוגר | לא ✗ | אין פעולה מונעת משמעותית |
| אלצהיימר בגיל צעיר | לא ✗ | אין טיפול מונע זמין כיום |
הכלל: מדווחים על Actionable Findings - ממצאים שבהם גילוי מוקדם מאפשר פעולה (מניעה, אבחון מוקדם, טיפול) שמשנה את המהלך הקליני.