כרומוזום Y ותורשה Y-linked
מאפייני כרומוזום Y
- גודל: קטן בהרבה מכרומוזום X
- תוכן גנטי מועט: רוב הכרומוזום לא מכיל גנים משמעותיים
- אזורים עיקריים:
- Pseudoautosomal region (PAR): אזור קטן המשותף עם כרומוזום X
- SRY gene: הגן הקריטי לקביעת המין הזכרי
- AZF regions (A,B,C): גנים ליצירת זרע (spermatogenesis)
גן SRY
- ממוקם קרוב לאזור הפסאודו־אוטוזומלי
- קובע התפתחות זכרית בעובר (אשכים וגניטליה זכרית)
- יוצר קסקדה של התפתחות זכרית
- חסר של SRY: גורם ל־XY עם פנוטיפ נקבי
- SRY על כרומוזום X (עקב crossing over): גורם ל־XX עם פנוטיפ זכרי
תורשה Y-linked
דגש: אין באמת מחלות Y-linked חשובות (לפחות לצורך הקורס שלנו).
- העברה: רק מאב לבן
- המחלה היחידה המשמעותית: ליקויי פריון/אזוספרמיה
- חסרים ב־AZF regions
- בעיה: איך עובר לדור הבא אם יש בעיה בפריון?
- תשובה: טיפולי פוריות (ביופסיה מהאשך, הזרקת זרע)
הערה על כהנים: ייתכן שיש מחקרים שבדקו כהנים ברחבי העולם ומצאו מרקרים של אותו אב קדמון בכרומוזום Y
כרומוזום X ו־X inactivation
X Inactivation (Lyonization)
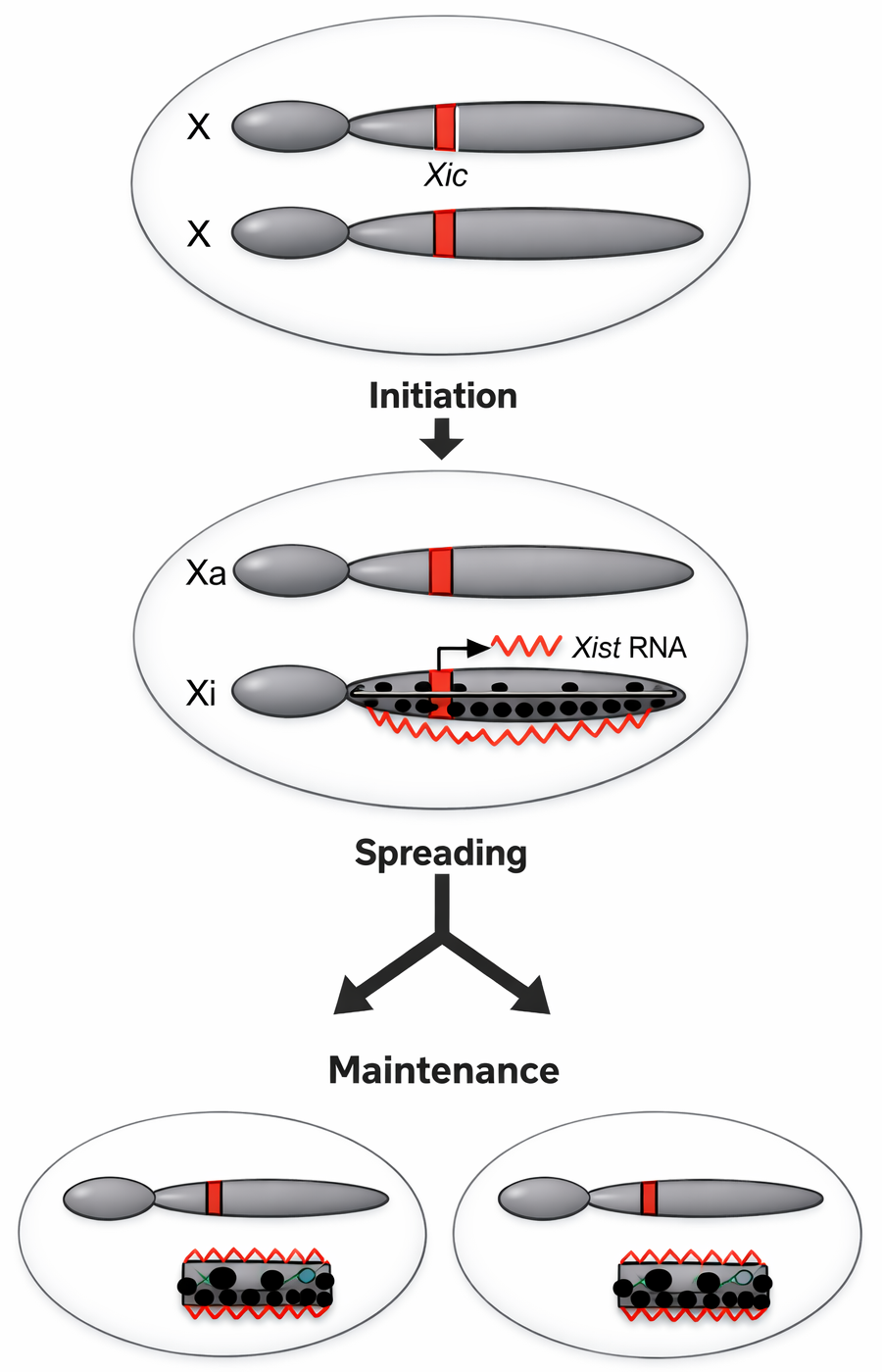
אטימולוגיה: Mary Lyon תיארה את זה לראשונה, לכן קוראים לזה גם Lyonization
המנגנון
- X inactivation center (XIC): מייצר long non-coding RNA בשם
XIST XIST RNA: עוטף את הכרומוזום X שעתיד להיות מושתק- התוצאה: Barr body - כרומוזום X דחוס וקומפקטי
- הזמן: קורה בשלב מוקדם של העובר (20-10 תאים)
- הכרומוזום המושתק נותר מושתק במהלך חלוקת התא (תאי הבת של תא מסוים ימשיכו את ההשתקה)
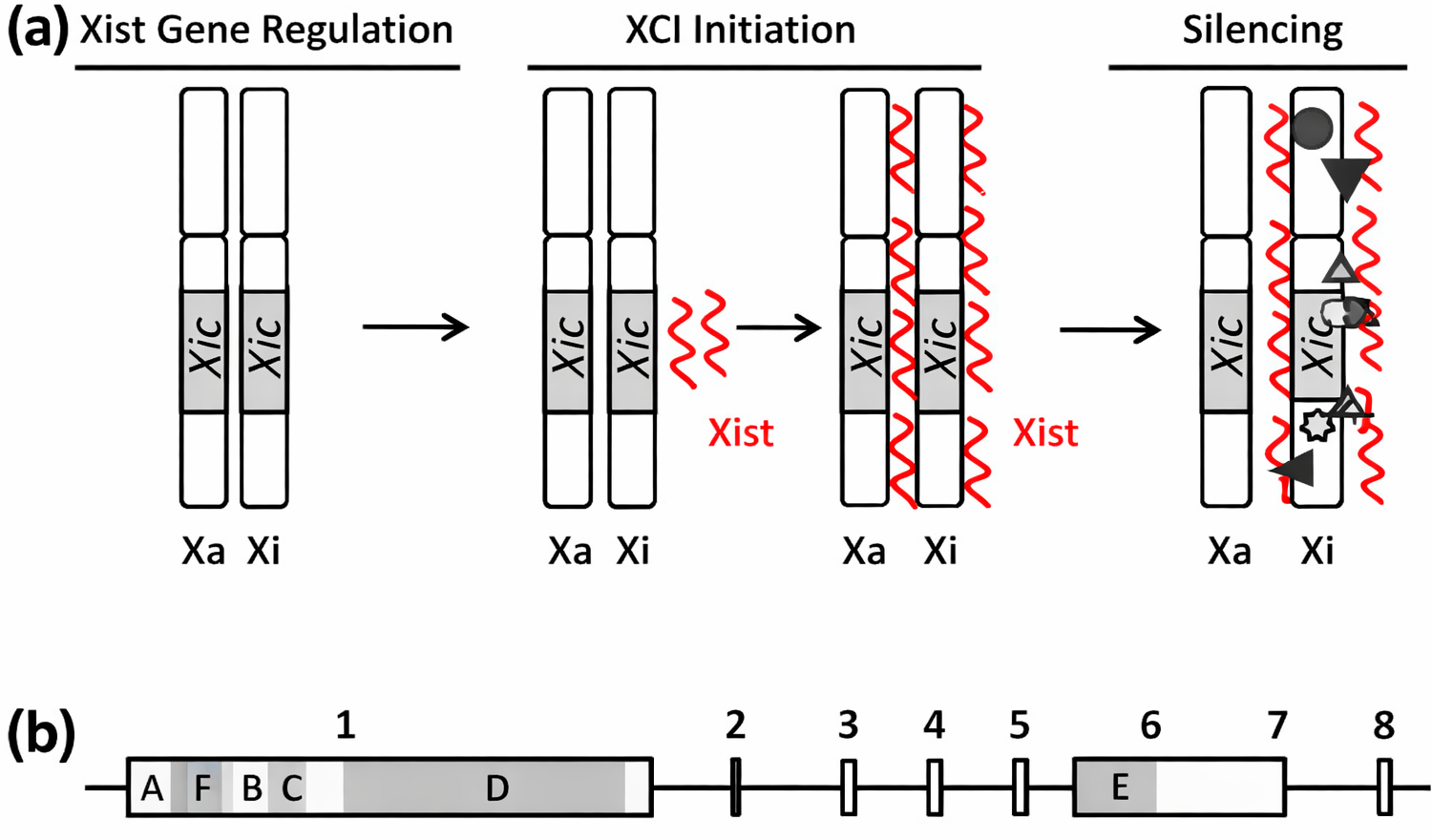
עקרונות חשובים
- רנדומליות: ההחלטה איזה X מושתק היא בדרך כלל אקראית
- Non-random (skewed) X inactivation:
- כאשר יש מוטציה ב־X אחד
- התאים עם ה־X הפגום מסתדרים פחות טוב
- לחץ אבולוציוני על התאים (לא על האדם!)
דוגמת החתולות: חתול עם 3 צבעים = נקבה, בגלל המוזאיקה מ־X inactivation.
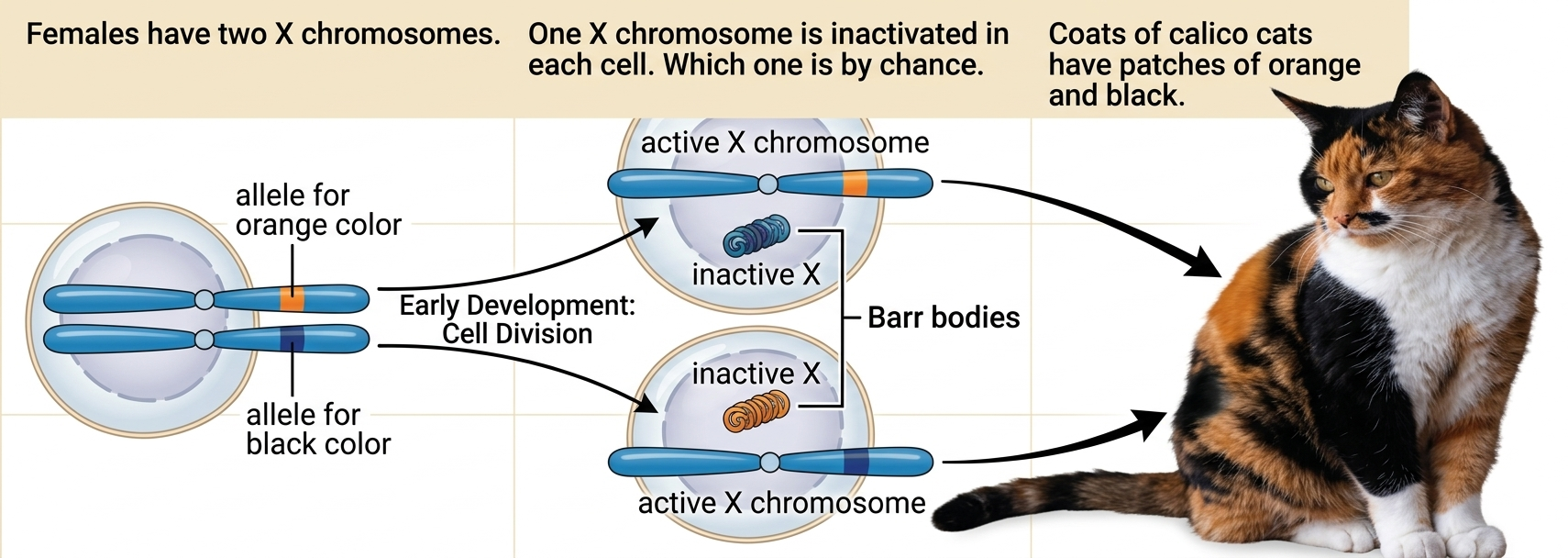
מספר לא תקין של כרומוזומי X
- Turner (45,X0): לא בר-חיים ברוב המקרים
- Klinefelter (47,XXY): משתיק X אחד
- הכלל: תמיד נשאר רק X אחד פעיל
מחלות X-linked Recessive
- אמא נשאית ← 50% בנים חולים, 50% בנות נשאיות
- אבא חולה ← כל הבנות נשאיות, כל הבנים בריאים
□ ─────┬────── ◐
│
┌──────────┬────┴─────┬──────┐
□ ○ ◐ ■ ──┬── ○
│
┌────┬───┴───┬────┐
◐ ◐ □ □
Duchenne Muscular Dystrophy (דוּשֶן)
הגן והחלבון
- גן DMD: מקודד לדיסטרופין
- דיסטרופין: מחבר בין היחידה המתכווצת לממברנת התא
- בלי דיסטרופין: התא לא יציב ומתפרק = דיסטרופיה
קליניקה (הוצגו תמונות)
- גיל 18 חודשים: איחור בהליכה
- Gower sign: “הילד מטפס על עצמו כדי לקום” - סימן לחולשת שרירי הירכיים
- Pseudohypertrophy: “נראה כאילו יש שרירים גדולים אבל זה החלפה לשומן”
- גיל 12: בכיסא גלגלים
- קרדיומיופתיה ובעיות נשימה
- CPK גבוה מאוד: עשרות אלפים, לא כמו מישהו שהלך לחדר כושר
דושן נמצא בבדיקות סקר גנטי לכל הזוגות בהריון
Duchenne vs Becker
- Duchenne: אין חלבון בכלל (deletion/frameshift)
- Becker: יש חלבון אבל קצר/לא תקין
- הבדל ניכר: בדושן - כיסא גלגלים בגיל 12, בבקר - עדיין הולכים בגיל 20
נשאיות
- יכולות להיות סימפטומטיות (בגלל X inactivation)
- CPK מעט גבוה
- סיכון לקרדיומיופתיה (בעיקר בדושן)
עיוורון צבעים
דגש: תכונה שכיחה שלא מקצרת חיים
- יותר שכיח בזכרים
- אם שכיח מאוד - יכול להיראות כמו אוטוזומלי רצסיבי
- דוגמה: אישה עם עיוורון צבעים = שני X פגומים
- כל הבנים שלה יהיו עיוורי צבעים
- הבנות - תלוי באבא
Fragile X Syndrome
המנגנון המיוחד
הערה: אחת מצורות התורשה הכי מעצבנות ומסובכות - גם X-linked, גם recessive, וגם repeats!
- גן FMR1: מכיל חזרות CGG
- תקין: עד 55 חזרות
Premutation: 200-55 חזרות- Full mutation: מעל 200 חזרות
Anticipation
- ככל שיש יותר חזרות, גדל הסיכוי להרחבה
- 100 חזרות: 100% סיכוי להרחבה מעל 200 בדור הבא
- הרחבה מתרחשת בעיקר במעבר מאם לילד
קליניקה
- פיגור שכלי (הסיבה השכיחה ביותר באוכלוסייה)
- אוטיזם, ADHD
- ראש גדול (macrocephaly)
- אורכידיזם (אשכים גדולים)
- פנים אופייניות: אוזניים גדולות, מצח גבוה, פנים משולשות
מחלות X-linked Dominant (דומיננטית בתאחיזה ל־X)
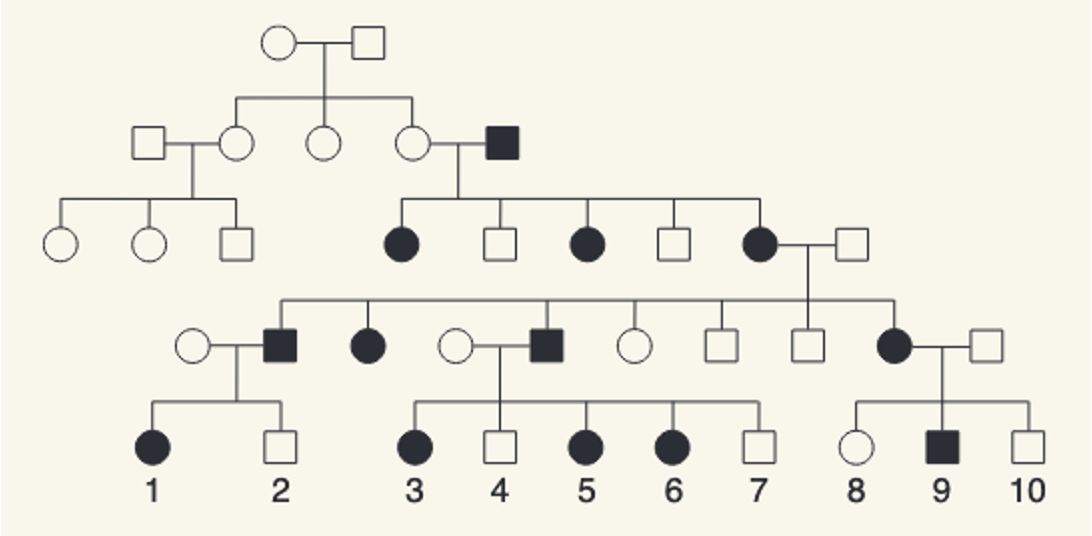
- גבר חולה ← כל הבנות חולות, כל הבנים בריאים
- אישה חולה ← 50% בנים חולים, 50% בנות חולות?
X-linked Hypophosphatemia
דוגמה מהקליניקה:
- ילדה עם רגליים לא סימטריות
- רככת (rickets) - עצמות רכות ומתעקמות
- “בורינג של הרגליים” - קשתות ברגליים
- פנים א-סימטריות
- טיפול: תוספת פוספט - הרגליים מסתדרות עם הזמן
Rett Syndrome
דגש: רואים רק בנות - Male lethal
מאפיינים קליניים
- התפתחות תקינה עד גיל שנה־שנה וחצי
- רגרסיה: “מה שהילדה ידעה - דיבור, הליכה - מאבדת”
- Hand-washing movements: תנועות ידיים אופייניות
- “לא purposeful use של הידיים”
- פיגור עמוק, אפילפסיה
הערה: המרצה ציינה שהיא מציגה את Rett Syndrome בגלל שהוא במבחן סקר גנטי לעוברים.
Incontinentia Pigmenti
סיפור מהקליניקה: תינוקת עם פריחה נוראית - חושבים הרפס, עושים workup מלא, אנטיביוטיקה, כלום לא עוזר
שלבי המחלה
- שלב ראשון: פריחה שלפוחייתית (נראה כמו הרפס)
- שלב שני: היפרקרטוזיס
- שלב שלישי: היפרפיגמנטציה בדרמטומים
מעניין: זה מוזאיקה בגלל X inactivation - לכן רואים את הפסים
X-linked Dominant Male-sparing
הגבר יכול לשרוד - זה ההבדל העיקרי.
PCDH19-related epilepsy
המנגנון המיוחד:
- פוגע רק בנקבות
- זכרים עם המוטציה - בריאים
- צריך את המוזאיקה כדי ליצור בעיה בחשמל במוח
- בזכר - כל התאים אותו דבר = אין אפילפסיה
- בנקבה - מוזאיקה = בעיה בחיבורים = אפילפסיה
Pseudoautosomal Region
SHOX Deficiency
- גן הנמצא גם ב־X וגם ב־Y
- מתנהג כמו אוטוזומלי דומיננטי
- גורם לקומה נמוכה עם קיצור גפיים
- הערה: מגיב טוב להורמון גדילה - מוסיף 9-8 ס״מ
- חסר SHOX גורם גם לקומה הנמוכה בטרנר
טיפים לזיהוי דפוסי תורשה
סיכום:
- X-linked recessive:
- בעיקר זכרים חולים
- עובר דרך נשאיות
- לא עובר מאב לבן
- אב חולה ← כל הבנות נשאיות (אם האם בריאה)
- X-linked dominant:
- גבר חולה = כל הבנות חולות, אף בן לא חולה (בנים מקבלים מהאב Y)
- אישה חולה (הטרוזיגוטית) = 50% בנים חולים, 50% בנות חולות
- איך לשלול X-linked:
- אם יש מעבר מאב לבן = לא X-linked
- “אם רואים אבא מעביר לבן - 100% לא X-linked!”
הערה כללית: במקרה של דומיננטי, כל נשאית היא גם חולה. אז אם נתון שאישה נשאית (להבנתי בדרך כלל מתכוונים בריאה ונשאית - אחרת היו כותבים חולה), כנראה שמדובר במקרה של X-linked recessive ולא דומיננטי.
חישובי בייס (Bayesian Analysis)
עקרונות בסיסיים
- מטרה: לדייק את חישובי הסיכון בגנטיקה באמצעות מידע נוסף
- שימוש: כאשר יש לנו מידע נוסף על הפרובנד שיכול לדייק סטטיסטית את הסיכון
מרכיבי החישוב הבייזיאני
-
Prior Probability (הסתברות מוקדמת)
- הסיכוי הבסיסי על סמך המידע הגנטי בלבד
- לדוגמה: אישה שאביה חולה במחלה X-linked רצסיבית - סיכוי של 50% להיות נשאית
-
Conditional Probability (הסתברות מותנית)
- מידע נוסף שמשפיע על ההסתברות
- לדוגמה: אם לאישה יש ארבעה בנים בריאים
-
Joint Probability (הסתברות משותפת)
- מכפלת ההסתברות המוקדמת בהסתברות המותנית:
-
Posterior Probability (הסתברות סופית)
- ההסתברות המחושבת לאחר שקלול כל המידע
דוגמה מפורטת - מחלה X-linked רצסיבית
נתונים:
- אישה שאביה חולה (בהכרח נשאית)
- יש לה ארבעה בנים בריאים
- מה הסיכוי שהבת שלה נשאית?
חישוב:
| סיטואציה | Prior | Conditional | Joint |
|---|---|---|---|
| האישה נשאית + הבת נשאית | 1/2 | $(1/2)^4 \times 1/2$ | 1/64 |
| האישה נשאית + הבת לא נשאית | 1/2 | $(1/2)^4 \times 1/2$ | 1/64 |
| האישה לא נשאית | 1/2 | 1 | 1/2 |
תוצאה:
- סיכוי שהבת נשאית: $\frac{1/64}{1/64 + 1/64 + 1/2} = \frac{1}{33}$
- סיכוי שהבת לא נשאית: $\frac{33}{34}$
דוגמה נוספת - מחלה דומיננטית עם חדירות חלקית
מחלת Cleft-hand/foot:
- מחלה דומיננטית
- חדירות (penetrance) של 70%
- 30% מהנשאים לא מפתחים קליניקה
חישוב לאדם ללא סימנים קליניים:
- סיכוי להיות נשא: $\frac{0.5 \times 0.3}{(0.5 \times 0.3) + (0.5 \times 1)} = \frac{3}{13}$
דוגמה מהתרגול
נתון אילן היוחסין הבא. שני האחים של הפרובנד סובלים ממחלת קנדי (צורת הורשה של X-link). מכיוון ששני האחים חולים אנחנו מינחים שאמה של הפרובנד נשאית אובליגטורית.
לפני שנודע לנו ששלושת הבנים של הפרובנד אינם חולים, היינו אומרים שהסבירות או הסיכון שהיא תהיה נשאית (Carrier) כמו אמה הוא 1/2. אבל יש כאן נתונים נוספים.
כיצד משתנה הסיכוי של הפרובנד להיות נשאית אם ניקח בחשבון את העובדה שיש לה שלושה בנים בריאים?
□ ─────┬────── ◐
│
┌──────────┼──────────┐
■ ■ prob->○ ──┬── □
│
┌──────────┼──────────┐
□ □ □
event Prior Conditional Joint
--------------------------------------------------------------------
Carrier (C) 1/2 (1/2)^3 = 1/8 (1/2)*(1/8) = 1/16
Non-carrier (¬C) 1/2 1 (1/2)*1 = 1/2
תוצאה:
- סיכוי שהפרובנד נשאית: $\frac{1/16}{1/16 + 1/2} = \frac{1}{9}$
\[P(\text{sick}) = P(C) \times P(\text{boy}) \times P(\text{sick} | C) = \frac{1}{9} \times \frac{1}{2} \times \frac{1}{2} = \frac{1}{36}\]שאלת המשך מהתרגולים למבחן - מה הסיכון לילד חולה במחלת קנדי בהריון הבא?
מיטוכונדריה ומחלות מיטוכונדריאליות
תפקוד המיטוכונדריה
- ייצור אנרגיה: מגלוקוז דרך גליקוליזיס ← פירובט ← מעגל קרבס ← שרשרת הובלת אלקטרונים
- Oxidative Phosphorylation: התהליך המרכזי לייצור ATP
- β-Oxidation: ייצור אנרגיה משומנים
DNA מיטוכונדריאלי - מאפיינים
מבנה ותכונות
- צורה: מעגלית
- גודל: קטן יחסית ל־DNA גרעיני
- מספר גנים- 37 גנים:
- 13 גנים מקודדים לחלבוני Oxidative Phosphorylation
- 22 גנים ל־Transfer RNA
- 2 גנים ל־Ribosomal RNA
- עותקים מרובים: הרבה עותקים בכל מיטוכונדריון
תורשה מיטוכונדריאלית
- Maternal Inheritance: תורשה אימהית בלבד
- האם מעבירה לכל ילדיה (בנים ובנות)
- גברים חולים לא מעבירים לילדיהם
- הזרע לא תורם DNA מיטוכונדריאלי (או תרומה זניחה)
מושגים חשובים
Replicative Segregation
- המיטוכונדריות מתחלקות באופן עצמאי מהתא
- חלוקה רנדומלית בין תאי הבת
Homoplasmy vs Heteroplasmy
- Homoplasmy: כל ה־DNA המיטוכונדריאלי בתא זהה
- Heteroplasmy: תערובת של DNA מיטוכונדריאלי תקין ומוטנטי
- אחוז ההטרופלזמיה משפיע על חומרת המחלה
Threshold Effect
- נדרש אחוז מינימלי של מיטוכונדריות פגועות לביטוי קליני
- הסף משתנה בין רקמות שונות
סוגי מוטציות מיטוכונדריאליות
1. Deletions
- Kearns-Sayre Syndrome:
- בדרך כלל de novo בביצית
- שיתוק שרירי עיניים
- הפרעות קצב לב
- בעיות ראייה
2. מוטציות ב־Transfer RNA או Ribosomal RNA
- פגיעה רחבה בתפקוד המיטוכונדריה
- ביטוי קליני חמור יותר
- MELAS: מחלה קשה עם אירועים מוחיים, אפילפסיה, לקטת גבוה
3. מוטציות בגנים המקודדים לחלבוני Electron Transport Chain
- 13 גנים מיטוכונדריאליים
- מאות גנים גרעיניים נוספים
ביטוי קליני של מחלות מיטוכונדריאליות
רקמות מועדות לפגיעה (צורכות הרבה ATP)
- מוח: אפילפסיה, אירועים דמויי stroke, פיגור
- שריר: חולשה, Ragged Red Fibers בביופסיה
- לב: קרדיומיופתיה, הפרעות הולכה
- עיניים: לקות ראייה, שיתוק שרירי עיניים
- אוזניים: לקות שמיעה
- כליות: פגיעה כלייתית
- כבד: פגיעה בתפקודי כבד
מאפיינים קליניים של מחלות מיטוכונדריאליות
- וריאביליות רבה: אפילו באותה משפחה
- גיל הופעה משתנה: מלידה ועד גיל מבוגר
- לקטת גבוה בדם: סימן אופייני למחלות מיטוכונדריאליות
- החמרה במצבי stress: מחלות חום, זיהומים
אתגרים באבחון
קשיים באיתור המוטציה
- הטרופלזמיה משתנה בין רקמות
- צורך בביופסיה מהרקמה הפגועה
- ריצוף גנומי רגיל לא תמיד מכסה DNA מיטוכונדריאלי
בעיות בייעוץ גנטי
- קושי לחזות חומרת המחלה בעובר
- הטרופלזמיה משתנה בין אחים
- סיכון שנות: 100-0%
טיפול במחלות מיטוכונדריאליות
- אין טיפול מרפא
- טיפול תומך בלבד
- ניסיונות להחדרת מיטוכונדריות בריאות - עדיין בשלבי מחקר
Three-Parent Baby
- טכניקה ניסיונית למניעת העברת מחלות מיטוכונדריאליות
- שילוב של:
- DNA גרעיני מהאם הביולוגית
- DNA גרעיני מהאב
- DNA מיטוכונדריאלי מתורמת ביצית
חשיבות קלינית
- מחלות מיטוכונדריאליות יכולות להסביר:
- תינוקות שמתמוטטים במחלת חום ראשונה
- מחלות רב־מערכתיות עם תמונה לא ברורה
- הזדקנות של רקמות (צבירת מוטציות עם הזמן)
שאלות תרגול מג׳ונרטות
- השבתת X (XIST, Barr bodies, Skewed inactivation)
- DMD vs BMD ונשאיות סימפטומטיות
- תורשה צמודת Y (גני SRY, AZF, ICSI)
- האזור הפסאודואוטוזומלי (SHOX)
- X-linked recessive ו־dominant (כולל male lethal - Rett, Incontinentia Pigmenti)
- Fragile X
- גנום מיטוכונדריאלי, הומופלסמיה/הטרופלסמיה, אפקט סף, סגרגציה מיטוטית
- סוגי מוטציות mtDNA (Kearns-Sayre, MERRF, MELAS)
- אנליזה בייסיאנית - עם obligate carrier, עם בנים בריאים, ועם חדירות חלקית (70%)
- אתגרים באבחון טרום-לידתי של מחלות מיטוכונדריאליות
שאלה 1: השבתת כרומוזום X - מנגנון מולקולרי
מהו התפקיד המרכזי של ה־RNA הלא־מקודד XIST בתהליך השבתת X?
- XIST מתועתק ומתרגם לחלבון רגולטורי שנקשר לפרומוטורים על כרומוזום X ומכבה אותם אחד־אחד
- XIST מפעיל באופן ישיר אנזימי DNA-methyltransferase שממתילים בעיקר את כרומוזום X הפעיל וכך מייצבים את הבחירה
- XIST נשאר מולקולת RNA שמצפה את כרומוזום X המושבת ומגייס קומפלקסי השתקה (כגון קומפלקסי כרומטין) ליצירת הטרוכרומטין
- XIST משמש כאתר התחלה לשכפול מוקדם של כרומוזום X הפעיל וכך יוצר הבדלי תזמון בין שני העותקים
פתרון
התשובה הנכונה היא (3).
XIST הוא RNA לא־מקודד (Non-coding RNA) שמקודד מתוך ה־XIC (X Inactivation Center) הממוקם ברצועה Xq11.2, באזור בגודל כ־800Kb. ה־XIST נשאר כמולקולת RNA - הוא לא מתורגם לחלבון - ונקשר ומצפה את כרומוזום X הלא־פעיל. לאחר מכן הוא מגייס קומפלקסי “silencing” כגון היסטונים, המובילים לקונדנסציה של הכרומוזום ליצירת הטרוכרומטין.
למה האחרות שגויות:
- (א) XIST לא מקודד לחלבון כלל - הוא פועל כ־RNA.
- (ב) XIST עצמו אינו אנזים מתלציה - הוא מגייס קומפלקסים אחרים לתפקיד זה.
- (ד) XIST לא קשור לשכפול מוקדם; להפך - כרומוזום X הלא־פעיל משתכפל באיחור (replicates last).
מקור: שקפים 16–18
שאלה 2: גופיפי Barr ומספר כרומוזומי X
אישה עם קריוטיפ 47,XXX - כמה גופיפי Barr נצפה למצוא בגרעיני התאים שלה?
- 1, כי תמיד מושבת רק כרומוזום X אחד ללא תלות במספר העותקים הכולל
- 2, כי מספר גופיפי Barr שווה למספר כרומוזומי X פחות אחד
- 3, כי בכל תא כל כרומוזום X עובר קונדנסציה לגופיף Barr בנפרד
- 0, כי כרומוזומי X עודפים אינם מושתקים אלא מפורקים במהלך חלוקת התא
פתרון
התשובה הנכונה היא (2).
הכלל הוא: מספר גופיפי Barr = מספר כרומוזומי X − 1. תא תקין צריך כרומוזום X פעיל אחד בלבד - כל השאר מושבתים והופכים לגופיפי Barr. באישה 47,XXX: שלושה כרומוזומי X − 1 = שני גופיפי Barr. גופיף ה־Barr הוא כרומוזום X מגובש (highly condensed) הנמצא בפריפריה של הגרעין, משתכפל מאוחר, ונשאר לא־פעיל בתאי הבת.
למה האחרות שגויות:
- (א) לא רק אחד מושבת - כל עותק מעבר לאחד מושבת.
- (ג) לא כולם מושבתים - תמיד נשאר אחד פעיל.
- (ד) העותקים לא עוברים פירוק אלא קונדנסציה להטרוכרומטין.
מקור: שקפים 17, 20–21
מדובר אגב בטריזומיית X - מצב שבו יש שלושה כרומוזומי X. בכתה דווקא למדנו לדעתי יותר על קליינפלטר (XXY) וטרנר (X0), אבל העיקרון של מספר גופיפי Barr נשאר זהה בכל מצב עם יותר מ־X אחד.
שאלה 3: תכונות השבתת X
איזה מהמשפטים הבאים לגבי השבתת X הוא נכון?
- ההשבתה מתרחשת בעיקר לאחר יצירת איברים, ולכן אינה יוצרת מוזאיקה משמעותית ברקמות
- ההשבתה מוחלטת: כל הגנים על X המושבת מושתקים ללא חריגים
- ההשבתה מתרחשת מוקדם, היא אקראית, וה־X שנבחר להשבתה נשאר לא־פעיל בכל תאי הבת של אותו שושלת תאים
- ההשבתה אינה אקראית בבני אדם: תמיד X אימהי מושבת ו־X אבהי נשאר פעיל
פתרון
התשובה הנכונה היא (3).
השבתת X מתרחשת מוקדם בהתפתחות העוברית, היא אקראית (Random), ואותו כרומוזום X שהושבת נשאר לא־פעיל בכל תאי הבת
(the same X chromosome remains inactive through further cell division).
בנוסף, כרומוזום X אבנורמלי מבנית נוטה להיות זה שמושבת - בגלל סלקציה משנית, שכן תאים עם חוסר איזון שורדים פחות טוב.
למה האחרות שגויות:
- (א) ההשבתה מתרחשת מוקדם מאוד - ולכן כל אישה היא מוזאיקה של שתי אוכלוסיות תאים.
- (ב) לא כל הגנים מושבתים - כ־50% מהגנים בקצה הדיסטלי של Xp חומקים מהשבתה, וגנים בודדים ב־Xq גם כן.
- (ד) ההשבתה אקראית ולא תלויה במקור ההורי.
מקור: שקפים 13, 19
שאלה 4: Skewed X Inactivation ומשמעות קלינית
נשאית של מוטציה בגן ה־DMD מפתחת חולשת שרירים וקרדיומיופתיה. מהו ההסבר הסביר לכך שנשאית הטרוזיגוטית מציגה תסמינים?
- מוטציה חדשה הופיעה ב־X התקין בכל תאי השריר ולכן נוצר מצב פונקציונלי של “שני עותקים פגומים”
- DMD מתנהגת כדומיננטית בנשים, ולכן כל נשאית צפויה לפתח תסמינים קבועים עם הגיל
- השבתת X מוטה: ברוב התאים הושבת דווקא ה־X התקין, ולכן העותק הפעיל ברוב התאים הוא זה שנושא את המוטציה
- הדיסטרופין המוטנטי פועל כ”חלבון דומיננטי-שלילי” ופוגע גם בתאים שמייצרים דיסטרופין תקין
פתרון
התשובה הנכונה היא (3).
בנשאיות DMD, כ־76% אינן מציגות תסמינים כלל, אך כ־19% מציגות חולשת שרירים ו־8% מפתחות קרדיומיופתיה מורחבת. ההסבר הוא Skewed X inactivation - כאשר באופן אקראי (או בסלקציה) רוב התאים השביתו דווקא את כרומוזום X התקין, והעותק הפעיל ברוב התאים הוא זה הנושא את המוטציה. ב־90% מהנשים ההשבתה קרובה ליחס 50:50, אך בקצוות ההתפלגות ייתכן יחס קיצוני שמוביל לביטוי קליני.
למה האחרות שגויות:
- (א) מוטציה שנייה ב־X השני אפשרית תאורטית אך נדירה מאוד - לא זה ההסבר השכיח.
- (ב) DMD היא רצסיבית צמודת X - לא דומיננטית חלקית.
- (ד) דיסטרופין פגום אינו רעיל - הבעיה היא חוסר דיסטרופין תקין, לא נוכחות חלבון מזיק.
מקור: שקפים 30–32
שאלה 5: DMD לעומת BMD - מנגנון מולקולרי
מה מסביר את ההבדל בחומרה הקלינית בין Duchenne ל־Becker Muscular Dystrophy, למרות ששתיהן נגרמות ממוטציות באותו גן?
- ב־DMD המוטציה לרוב פוגעת בפרומוטור ולכן אין תעתוק; ב־BMD המוטציה אקסונית ולכן יש תעתוק חלקי
- ב־DMD מוטציות רבות גורמות ל־frameshift/Stop וחלבון לא־תפקודי; ב־BMD מוטציות In-frame מאפשרות חלבון קצר אך בעל תפקוד חלקי
- ב־DMD אין כלל יצירת mRNA; ב־BMD יש mRNA אך אין כלל תרגום לחלבון
- ההבדל נקבע בעיקר לפי מספר האקסונים שנמחקו, עם סף קבוע שמפריד בין DMD ל־BMD
פתרון
התשובה הנכונה היא (2).
ב־DMD המוטציות (מחיקות גדולות, מוטציות Stop, מוטציות שחבור) פוגעות באופן חמור בחלבון הדיסטרופין - לרוב שינוי מסגרת הקריאה (frameshift) שמוביל לחלבון מקוצר ולא־תפקודי. ב־BMD, דלציות או דופליקציות ה”שומרות על מסגרת הקריאה” (in-frame) מייצרות חלבון שקצר מהרגיל אך עדיין פונקציונלי חלקית. לכן ב־DMD: הופעה בגיל 3–5, כיסא גלגלים עד גיל 12, מוות עד גיל 20. ב־BMD: הליכה עד גיל 16, פנוטיפ משתנה.
למה האחרות שגויות:
- (א) המוטציות בשני המצבים הן באקסונים ולא בהכרח בפרומוטור - ההבדל הוא בסוג המוטציה ולא במיקומה.
- (ג) גם ב־DMD מיוצר mRNA - הבעיה היא בחלבון שנוצר ממנו.
- (ד) לא מספר האקסונים שנמחקו קובע אלא האם המחיקה שומרת על מסגרת הקריאה.
מקור: שקפים 27–29
שאלה 6: תורשה צמודת Y - מאפיינים
מהם המאפיינים המבדילים של תורשה צמודת כרומוזום Y?
- התכונה עוברת מאב לכל ילדיו (בנים ובנות), כי Y מבצע רקומבינציה מלאה עם X
- רק גברים מושפעים, ואב חולה מעביר את התכונה לכל בניו אך לאף אחת מבנותיו
- רק גברים מושפעים, אך ההעברה תלויה בכך שהאם נשאית של אלל מקביל על כרומוזום X
- נשים וגברים מושפעים בשווה, כי הגנים על Y זהים לאלה שעל X
פתרון
התשובה הנכונה היא (2).
בתורשה צמודת Y (Y-linked), אב נושא העברה בתכונה מעביר אותה לכל בניו (שמקבלים ממנו Y) אך לאף אחת מבנותיו (שמקבלות ממנו X). לכן רק גברים מושפעים, ודפוס ההעברה הוא אבהי בלבד מדור לדור. דוגמה קלאסית: Hypertrichosis (שיעור יתר באוזניים - Hairy ear syndrome).
למה האחרות שגויות:
- (א) כרומוזום Y עובר לבנים בלבד; רקומבינציה עם X מתרחשת רק באזור הפסאודואוטוזומלי.
- (ג) נשים אינן יכולות להיות נשאיות של מוטציה צמודת Y - אין להן כרומוזום Y.
- (ד) כרומוזום Y מכיל מעט גנים ייחודיים שאינם זהים לאלה שעל X.
מקור: שקפים 7–10
שאלה 7: גנים על כרומוזום Y
לגבר אובחנה אזואוספרמיה והבדיקה מראה מיקרו־דלציה באזור AZFc על כרומוזום Y. מה נכון לגבי מצב זה?
- אם ייוולד בן באמצעות ICSI - הוא לא יירש את הדלציה כי AZFc אינו באזור PAR
- אם ייוולד בן באמצעות ICSI - הוא צפוי לרשת את כרומוזום ה־Y עם הדלציה ועלול לסבול מבעיות פוריות דומות
- הדלציה מצביעה בהכרח על פגיעה ב־SRY ולכן ייתכן שהקריוטיפ אינו XY תקין
- AZFc אינו קשור ליצירת זרע ולכן הממצא אינו רלוונטי לייעוץ גנטי
פתרון
התשובה הנכונה היא (2).
אזורי AZF (AZFa, AZFb, AZFc) על כרומוזום Y מכילים גנים חיוניים ליצירת זרע (Spermatogenesis). מיקרו־דלציה באזור AZFc היא סיבה גנטית שכיחה לאזואוספרמיה. אם הזוג יבצע ICSI (הזרקת זרע תוך־ציטופלזמטית) וייוולד בן - הבן יקבל את כרומוזום Y של אביו עם הדלציה, ולכן צפוי לסבול מבעיות פוריות דומות. זהו מידע קריטי בייעוץ גנטי לפני הליך ICSI.
למה האחרות שגויות:
- (א) הדלציה נמצאת על כרומוזום Y ותעבור לכל הבנים - היא חלק מהכרומוזום עצמו.
- (ג) SRY הוא גן נפרד מ־AZF; דלציה ב־AZFc אינה פוגעת ב־SRY.
- (ד) אזורי AZF אחראים ישירות ליצירת זרע - לא לגובה או מבנה גוף.
מקור: שקפים 10–11
שאלה 8: האזור הפסאודואוטוזומלי (PAR)
מהי המשמעות הקלינית של העובדה שגן SHOX ממוקם באזור הפסאודואוטוזומלי (PAR1)?
- מוטציה ב־SHOX תתבטא רק בזכרים כי הגן נמצא על כרומוזום Y בלבד
- בגלל מיקום ב־PAR, דפוס ההורשה דומה לאוטוזומלי ויכול לעבור מכל הורה לכל ילד/ה ללא תלות במין
- מכיוון שהגן על X בלבד, דפוס ההורשה הוא X-linked recessive קלאסי
- הגן חומק מהשבתת X אך עדיין מועבר רק מאם לבנות ולכן נשאר “צמוד X”
פתרון
התשובה הנכונה היא (2).
האזור הפסאודואוטוזומלי (PAR) הוא אזור בקצוות כרומוזומי X ו־Y שבו מתרחשת רקומבינציה (crossing over) בין X ל־Y במהלך מיוזה. גנים באזור זה קיימים הן על X והן על Y, ולכן דפוס ההורשה שלהם דומה לאוטוזומלי. חוסר (Deficiency) בגן SHOX גורם לקומה נמוכה ולעיוות מדלונג (Madelung deformity) - ויכול להופיע גם בגברים וגם בנשים.
למה האחרות שגויות:
- (א) SHOX נמצא גם על X וגם על Y - לא רק על Y.
- (ג) מכיוון שהגן קיים גם על Y, זו לא תורשה צמודת X קלאסית.
- (ד) ההורשה היא פסאודואוטוזומלית - לא צמודת X בלבד.
מקור: שקפים 59–61
שאלה 9: זיהוי דפוס תורשה - X-linked recessive
במשפחה: סבא (דור I) חולה בהמופיליה. בתו (דור II) בריאה ונשואה לגבר בריא. לזוג 4 בנים בריאים ובת אחת. מה הסיכון שהבת (דור III) היא נשאית?
- 100%, כי כל בת של נשאית היא נשאית בהכרח
- 50%, כי האם היא נשאית ודאית ולכן לכל בת יש הסתברות 1/2 לרשת את ה־X הפגום
- 25%, כי יש להכפיל סיכוי שהאם נשאית (1/2) וסיכוי שהבת יורשת את האלל (1/2)
- 0%, כי ארבעה בנים בריאים שוללים שהאם נשאית
פתרון
התשובה הנכונה היא (2).
הסבא (I) חולה בהמופיליה (X-linked recessive) ← הגנוטיפ שלו X^h Y. הוא מעביר את כרומוזום X^h שלו לכל בנותיו ← בתו (II) היא בהכרח נשאית (Obligate carrier) עם גנוטיפ X^H X^h. מכיוון שהאם נשאית ודאית, כל בת שלה בסיכון 50% לקבל את X^h ולהיות נשאית.
העובדה ש־4 בנים בריאים אינה שוללת נשאות אצל האם - זה מידע שניתן להשתמש בו באנליזה בייסיאנית לעדכון הסיכון, אך ההסתברות ה־Prior של האם להיות נשאית היא 100% (Obligate carrier).
למה האחרות שגויות:
- (א) הבת בסיכון 50% ולא 100% - האם מעבירה לכל בת אחד משני כרומוזומי X שלה באקראי.
- (ג) האם היא נשאית ודאית (100%), לא 50%.
- (ד) בנים בריאים אינם שוללים נשאות - הם פשוט ירשו את X התקין.
מקור: שקפים 56, 62
שאלה 10: מאפייני X-linked dominant
איזו מהתצפיות הבאות עקבית עם דפוס תורשה X-linked dominant?
- אב חולה ואם בריאה - כל הבנים חולים וכל הבנות בריאות
- אב חולה ואם בריאה - כל הבנות חולות, אף אחד מהבנים לא חולה
- אם חולה ואב בריא - כל הילדים חולים ללא תלות במין
- שני ההורים בריאים - 25% מהבנים ו־25% מהבנות חולים בכל הריון
פתרון
התשובה הנכונה היא (2).
ב־X-linked dominant, אב חולה (X^A Y) מעביר את כרומוזום X שלו (הנושא את המוטציה) לכל בנותיו ← כל הבנות חולות. לבנים הוא מעביר כרומוזום Y ← אף בן לא חולה. זהו סימן היכר מובהק:
Affected male with unaffected partner has no affected sons and no unaffected daughters.
כאשר האם היא החולה (X^A X), לכל ילד/ה סיכוי של 50% לחלות - גם בנים וגם בנות. נשים חולות שכיחות פי שניים מגברים חולים, אך בדרך כלל עם תסמינים קלים יותר (תלוי ב־skewing של X).
למה האחרות שגויות:
- (א) הפוך - הבנים מקבלים Y מהאב ולכן לא חולים; הבנות מקבלות X מהאב ולכן חולות.
- (ג) אם חולה מעבירה 50% סיכון לכל ילד - לא 100%.
- (ד) שני הורים בריאים ← ייתכן רק מוטציה de novo.
מקור: שקפים 37, 57
שאלה 11: X-linked dominant lethal בזכרים
תסמונת רט (Rett Syndrome) נגרמת ממוטציה בגן MECP2 על כרומוזום X. מדוע תסמונת רט מופיעה כמעט אך ורק בבנות?
- MECP2 מבוטא רק כשיש שני כרומוזומי X, ולכן בזכרים הוא שקט לגמרי
- בזכרים אין עותק X נוסף לפיצוי; מוטציה ב־X היחיד גורמת לרוב לפנוטיפ חמור שאינו מתיישב עם חיים
- המוטציה אינה יכולה לעבור לזכרים כי MECP2 נמצא באזור שלא עובר רקומבינציה עם Y
- MECP2 נמצא למעשה על כרומוזום Y, והמחלה בבנות נובעת מטרנסלוקציה נדירה בלבד
פתרון
התשובה הנכונה היא (2).
תסמונת רט היא X-linked dominant disorder שהיא lethal בזכרים. בנות עם מוטציה ב־MECP2 שורדות כי יש להן כרומוזום X שני תקין - בחלק מהתאים X התקין פעיל ומפצה. בזכרים, כרומוזום X היחיד נושא את המוטציה ← אין פיצוי ← פנוטיפ חמור מאוד (אנצפלופתיה ניאונטלית חמורה) שבדרך כלל גורם למוות מוקדם. הקליניקה אצל בנות: התפתחות נורמלית ב־6–18 חודשים הראשונים, ואז סטגנציה ורגרסיה מהירה בדיבור ובמוטוריקה, עם תנועות ידיים סטריאוטיפיות.
למה האחרות שגויות:
- (א) MECP2 מבוטא גם בזכרים - הבעיה היא שאין להם עותק תקין שיפצה.
- (ג) המוטציה יכולה לעבור לזכרים - אלא שהם לא שורדים.
- (ד) MECP2 ממוקם על כרומוזום X, לא Y.
מקור: שקפים 42–44
שאלה 12: Fragile X - מנגנון
מהו המנגנון המולקולרי בתסמונת Fragile X?
- מחיקה מלאה של גן FMR1 שמונעת יצירת mRNA וגורמת להיעדר מוחלט של FMRP
- התארכות חזרות CGG ב־FMR1 שמעל סף מסוים גורמת למתילציה/השתקה ולהיעדר FMRP
- מוטציית Missense ב־FMR1 שמייצרת חלבון FMRP רעיל המצטבר במוח
- טרנסלוקציה של FMR1 לאוטוזום שגורמת לביטוי יתר של FMRP
פתרון
התשובה הנכונה היא (2).
תסמונת Fragile X נגרמת מהתארכות חזרות CGG ב־5’UTR של גן FMR1 על כרומוזום X. כאשר מספר החזרות חוצה סף מסוים (Full mutation, >200 חזרות), הגן עובר מתילציה ומושתק ← אין ייצור חלבון FMRP ← פיגור שכלי. התורשה היא X-linked, עם מנגנון של Repeats. זהו הגורם התורשתי השכיח ביותר לפיגור שכלי/עיכוב התפתחותי. מאפיינים קליניים: מקרוצפליה ומקרו־אורכידיזם.
למה האחרות שגויות:
- (א) המנגנון אינו מחיקה אלא התארכות חזרות שמובילה להשתקה אפיגנטית.
- (ג) הבעיה היא חוסר חלבון - לא חלבון רעיל.
- (ד) אין טרנסלוקציה - הגן נשאר על כרומוזום X.
מקור: שקפים 50–52
שאלה 13: הגנום המיטוכונדריאלי - מאפיינים
כמה גנים מכיל הגנום המיטוכונדריאלי, ומה הם מקודדים?
- ~1,500 גנים המקודדים את רוב חלבוני המיטוכונדריה כולל אנזימי קרבס וחלבוני ETC
- 37 גנים: 2 rRNAs, 22 tRNAs, ו־13 פוליפפטידים שהם תת־יחידות של OXPHOS
- 13 גנים בלבד המקודדים רק לקומפלקסים I ו־II של שרשרת הנשימה
- ~80 גנים המקודדים את כל תתי-היחידות של חמשת הקומפלקסים של שרשרת האלקטרונים
פתרון
התשובה הנכונה היא (2).
הגנום המיטוכונדריאלי הוא כרומוזום מעגלי בגודל כ־16.5Kb, הממוקם בתוך האברון המיטוכונדריאלי (לא בגרעין). הוא מכיל 37 גנים: 2 ribosomal RNAs, 22 transfer RNAs, ו־13 פוליפפטידים שהם תת־יחידות של OXPHOS. חשוב - כ־80 חלבונים מעורבים בקומפלקס OXPHOS, אך רק 13 מקודדים ע”י mtDNA. שאר כ־1,500 חלבוני המיטוכונדריה מקודדים ע”י ה־DNA הגרעיני.
למה האחרות שגויות:
- (א) 1,500 חלבוני מיטוכונדריה מקודדים ע”י DNA גרעיני - לא מיטוכונדריאלי.
- (ג) 13 הפוליפפטידים הם תת־יחידות של כמה קומפלקסים - לא רק I ו־II.
- (ד) 80 חלבונים מעורבים ב־OXPHOS, אך רק 13 מהם מקודדים ב־mtDNA.
מקור: שקפים 79–80
שאלה 14: תורשה מיטוכונדריאלית - מנגנון ההורשה האימהית
מדוע mtDNA עובר בתורשה אימהית בלבד?
- ה־mtDNA האבהי מפורק עוד לפני ההפריה ולכן אינו נכנס כלל לזיגוטה
- מיטוכונדריות הזרע נכנסות, אך מסולקות/מפורקות לאחר ההפריה ולכן mtDNA בזיגוטה מקורו בביצית
- לזרע אין מיטוכונדריות ולכן אין לו mtDNA להעביר
- ה־mtDNA האבהי משתלב בגרעין העובר ולכן אינו נשמר כאברוני
פתרון
התשובה הנכונה היא (2).
לזרע יש מיטוכונדריות (בחלק האמצעי), אך הן נכנסות לביצית ומסולקות מהציטופלזמה של העובר בשלב מוקדם. כך שכל המיטוכונדריות בזיגוטה - ולכן כל ה־mtDNA - מגיעות מהביצית (האם). ביצית בוגרת מכילה יותר מ־100,000 עותקי mtDNA, שמהווים כשליש מסך ה־DNA בתא. לכן: כל הילדים של אם נושאת מוטציה מיטוכונדריאלית יירשו אותה - בנים ובנות, אך רק בנותיה יעבירו הלאה.
למה האחרות שגויות:
- (א) הפירוק מתרחש לאחר כניסה לביצית, לא לפני ההפריה.
- (ג) לזרע יש מיטוכונדריות - הן ממוקמות בצוואר הזרע ומספקות אנרגיה לתנועה.
- (ד) mtDNA אבהי לא משתלב בגרעין - הוא פשוט מפורק.
מקור: שקפים 82–83
שאלה 15: הומופלסמיה והטרופלסמיה
מהו ההבדל בין הומופלסמיה להטרופלסמיה, ומדוע הוא משמעותי קלינית?
- הומופלסמיה: כל עותקי mtDNA זהים; הטרופלסמיה: תערובת של mtDNA תקין ומוטנטי באותו תא
- הומופלסמיה: לכל מיטוכונדריה אותו מספר עותקים; הטרופלסמיה: שונות במספר העותקים בין מיטוכונדריות
- הומופלסמיה: קיימות רק מיטוכונדריות תקינות; הטרופלסמיה: אין מיטוכונדריות בתא
- הומופלסמיה: כל רקמות הגוף זהות; הטרופלסמיה: רקמות שונות מכילות “מיטוכונדריה ממינים שונים”
פתרון
התשובה הנכונה היא (1).
הומופלסמיה (Homoplasmy) = כל עותקי ה־mtDNA בתא (ובמיטוכונדריות) זהים - כולם Wild type או כולם מוטנטיים. הטרופלסמיה (Heteroplasmy) = קיום של עותקי mtDNA תקינים ומוטנטיים באותו תא, ואפילו באותה מיטוכונדריה בודדת.
המשמעות הקלינית: כאשר מוטציה חדשה ב־mtDNA נוצרת, היא בהתחלה קיימת במולקולה אחת בלבד - ונוצר מצב של הטרופלסמיה. באמצעות חלוקות תא אקראיות (Replicative segregation), יחס המוטנטי/תקין יכול להשתנות.
למה האחרות שגויות:
- (ב) ההבדל לא במספר עותקים אלא ברצף (מוטנטי מול תקין).
- (ג) הטרופלסמיה אינה חוסר מיטוכונדריות - אלא תערובת של עותקים שונים.
- (ד) המונחים מתייחסים לתוכן mtDNA בתוך תאים - לא להבדלים בין רקמות (למרות שרקמות שונות יכולות להיות בדרגות הטרופלסמיה שונות).
מקור: שקפים 89–90
שאלה 16: אפקט הסף במחלות מיטוכונדריאליות
מדוע תסמינים של מחלות מיטוכונדריאליות מופיעים בדרך כלל קודם ברקמת מוח, לב ושרירי שלד?
- ברקמות אלו יש מעט מיטוכונדריות, ולכן גם שינוי קטן גורם לכשל מיידי
- רקמות אלו תלויות במיוחד ב־OXPHOS ולכן סף הפגיעה הקלינית נמוך יותר ביחס הטרופלסמיה
- רקמות אלו מתחלקות מהר ולכן מצטברות בהן מוטציות mtDNA בקצב הגבוה ביותר
- רקמות אלו חשופות יותר לנזקים סביבתיים חיצוניים ולכן mtDNA נפגע בעיקר מהסביבה
פתרון
התשובה הנכונה היא (2).
אפקט הסף (Threshold effect): מספר מינימלי של עותקי mtDNA מוטנטיים נדרש לפני שנוצרת הפרעה בזרחון חמצוני ומופיעים סימנים קליניים. רקמות שתלויות מאוד במטבוליזם אוקסידטיבי - מוח, לב, שרירי שלד, רשתית, וטובולי כליה - פגיעות במיוחד. הסף ברקמות אלו נמוך יותר, כלומר גם אחוז נמוך יחסית של mtDNA מוטנטי יכול לגרום לתפקוד לקוי ולתסמינים.
למה האחרות שגויות:
- (א) למוח ולב יש דווקא הרבה מיטוכונדריות - הבעיה היא התלות הגבוהה במטבוליזם אוקסידטיבי.
- (ג) נוירונים ומיוציטים בלב מתחלקים לעיתים נדירות - אך עדיין פגיעים.
- (ד) הנזק המרכזי הוא מהמוטציות ב־mtDNA עצמו ומשינויי היחס הטרופלסמי, לא מגורמים סביבתיים חיצוניים.
מקור: שקף 91
שאלה 17: סגרגציה מיטוטית
כיצד מסבירה הסגרגציה המיטוטית (Mitotic segregation) את השונות הקלינית בתוך אותו חולה לאורך זמן?
- בכל חלוקת תא נוצרת בהכרח מוטציה חדשה ב־mtDNA ולכן המחלה תמיד מחמירה בקצב קבוע
- חלוקת מיטוכונדריות אקראית בין תאי בת משנה את יחס מוטנטי/תקין ברקמות שונות; רקמה יכולה לחצות סף ולהופיע סימפטום חדש
- הסגרגציה מכוונת “לתקן” מוטציות ולכן עם הזמן היחס המוטנטי תמיד יורד
- הסגרגציה מתייחסת רק לחלוקת כרומוזומים גרעיניים ולכן אין לה קשר ל־mtDNA
פתרון
התשובה הנכונה היא (2).
Mitotic segregation - חלוקה אקראית של אברוני המיטוכונדריה בין תאי הבת בזמן חלוקת תא. כתוצאה מכך, היחס בין mtDNA מוטנטי לתקין יכול להשתנות לאורך זמן ברקמות שונות. רקמות עם קצב חלוקת תאים גבוה חשופות במיוחד לשינויים ביחס ההטרופלסמי. אם היחס חוצה את הסף (Threshold) ברקמה מסוימת - יופיעו תסמינים חדשים. זה מסביר את השונות הקלינית התלויית-גיל ותלויית-רקמה שמאפיינת מחלות מיטוכונדריאליות.
למה האחרות שגויות:
- (א) ההסבר המרכזי הוא שינוי ביחס הקיים - לא יצירת מוטציות חדשות (אם כי קצב המוטציות ב־mtDNA גבוה פי 6–17 מ־DNA גרעיני).
- (ג) הסגרגציה אקראית ואינה מכוונת לתיקון - היא יכולה לשפר או להחמיר.
- (ד) הסגרגציה המיטוטית כאן מתייחסת לחלוקת מיטוכונדריות - לא לכרומוזומים גרעיניים.
מקור: שקפים 92–94
שאלה 18: סוגי מוטציות ב־mtDNA
מהם שלושת סוגי המוטציות העיקריים ב־mtDNA?
- מוטציות נקודתיות ב־tRNA/rRNA, סידורים מחדש (מחיקות/כפילויות), ומוטציות missense בגנים של OXPHOS
- טרנסלוקציות בין mtDNA לגרעין, אינוורזיות, ומוטציות בפרומוטור מיטוכונדריאלי
- מחיקה של כל mtDNA, שכפול יתר של mtDNA, ומוטציות בגרעין בגני ETC
- מוטציות nonsense בלבד, מחיקות rRNA, והחדרת טרנספוזונים ל־mtDNA
פתרון
התשובה הנכונה היא (1).
שלושת סוגי המוטציות העיקריים ב־mtDNA:
-
סידורים מחדש (Rearrangements) של mtDNA המובילים למחיקות/כפילויות - למשל תסמונת Kearns-Sayre הנגרמת ממחיקה גדולה ב־mtDNA, לרוב sporadically מביצית של אם בריאה.
-
מוטציות נקודתיות בגני tRNA ו־rRNA הפוגעות בסינתזת חלבונים מיטוכונדריאלית - נחשבות “חמורות” יותר כי הן משפיעות על תרגום כל החלבונים. דוגמאות: MERRF ו־MELAS.
-
מוטציות Missense ב־13 הגנים הקשורים לזרחון חמצוני.
למה האחרות שגויות:
- (ב) אין טרנסלוקציות או אינוורזיות ב־mtDNA - אלו מושגים של כרומוזומים גרעיניים.
- (ג) מחיקת mtDNA שלם אינה אפשרית (התא יידרדר), ומוטציות בגרעין הן קטגוריה נפרדת.
- (ד) אין טרנספוזונים ב־mtDNA, והמוטציות אינן מוגבלות ל־Nonsense בלבד.
מקור: שקפים 96–101
שאלה 19: אנליזה בייסיאנית - עקרון
בעץ משפחה של מחלה X-linked recessive, II-1 היא בת של אב חולה (obligate carrier). ל־II-1 יש 5 בנים בריאים. מהו הסיכון הפוסטריורי של III-6 להיות נשאית?
Gen I ■ ──┬── ○
│
Gen II ◐ ──┬── □
┌────┬────┼────┬────┬────┐
Gen III □ □ □ □ □ ○
III-6
- 1/2, כי II-1 נשאית ודאית ולכן לכל בת שלה סיכון 50% להיות נשאית
- 1/64, כי מכפילים 1/2 עבור כל בן בריא ואז מכפילים שוב עבור הבת
- 1/34, כי בייס “מוריד” את סיכוי II-1 להיות נשאית ולכן גם הבת יורדת מתחת ל־1/2
- לא ניתן לחשב ללא בדיקה מולקולרית כי בנים בריאים תמיד משנים את הסיכון באופן לא ידוע
פתרון
התשובה הנכונה היא (1).
II-1 היא בת של אב חולה ← היא obligate carrier (prior = 1, לא 1/2). הסיבה: אב חולה (X^h Y) מעביר את X^h לכל בנותיו ← II-1 בהכרח קיבלה X^h מאביה ולכן היא נשאית ודאית. העובדה ש־5 בנים בריאים - מידע תנאי (conditional) - לא משנה את הסטטוס שלה כנשאית ודאית.
לכן, כל בת של II-1 בסיכון 1/2 לקבל את X^h = 50% סיכון להיות נשאית.
שימו לב: אנליזה בייסיאנית הייתה רלוונטית אילו ה־Prior של II-1 להיות נשאית היה פחות מ־1 (כמו במקרה שהאם שלה נשאית ב־50% סיכוי). במקרה כזה, בנים בריאים היו “מורידים” את הסיכון.
למה האחרות שגויות:
- (ב) 1/64 יהיה רלוונטי אם ה־Prior של II-1 היה 1/2 ולא 1 - כלומר אם היא לא הייתה obligate carrier.
- (ג) התשובה מנסה ליישם Bayes אך מגיעה למסקנה זהה לתשובה א - II-1 היא נשאית ודאית.
- (ד) הסיכון ניתן לחישוב על סמך עץ המשפחה בלבד.
מקור: שקפים 62, 68–72
שאלה 20: אנליזה בייסיאנית עם בנים בריאים
ל־II-2 Prior של 1/2 להיות נשאית, ויש לה 5 בנים בריאים. מהו הסיכון הפוסטריורי של II-2 להיות נשאית?
Gen I □ ──┬── ◐
│
Gen II ○ ──┬── □
┌────┬────┼────┬────┐
Gen III □ □ □ □ □
- 1/2, כי מידע על בנים בריאים לא משנה סיכון לנשאות
- 1/33, כי מעדכנים לפי הסתברות של (1/2)^5 לקבל 5 בנים בריאים אם היא נשאית
- 1/64, כי זהו הסיכוי המצטבר ל־5 בנים בריאים אם היא נשאית ולכן זהו הפוסטריור
- 0, כי רצף של בנים בריאים שולל נשאות באופן דטרמיניסטי
פתרון
התשובה הנכונה היא (2).
אנליזה בייסיאנית:
| נשאית | לא נשאית | |
|---|---|---|
| Prior | 1/2 | 1/2 |
| Conditional (בן 1 בריא) | 1/2 | 1 |
| Conditional (בן 2 בריא) | 1/2 | 1 |
| Conditional (בן 3 בריא) | 1/2 | 1 |
| Conditional (בן 4 בריא) | 1/2 | 1 |
| Conditional (בן 5 בריא) | 1/2 | 1 |
| Joint | 1/2 × (1/2)^5 = 1/64 | 1/2 × 1 = 1/2 |
| Posterior | (1/64) / (1/64 + 1/2) = 1/33 | 32/33 |
כל בן בריא מוריד את הסיכון - כי אם II-2 נשאית, הסיכוי שכל 5 הבנים יהיו בריאים הוא (1/2)^5 = 1/32. הסיכון הפוסטריורי של II-2 להיות נשאית הוא 1/33.
למה האחרות שגויות:
- (א) בנים בריאים כן מעדכנים את הסיכון - זהו כל הרעיון של Bayes.
- (ג) 1/64 הוא ה־Joint ולא ה־Posterior - צריך לחלק בסכום ה־Joints.
- (ד) בנים בריאים מפחיתים סיכון אך לא שוללים לחלוטין - תמיד נשאר סיכוי שסטטיסטית “התמזל מזלה”.
מקור: שקפים 68–72
שאלה 21: אנליזה בייסיאנית עם חדירות חלקית
במחלה אוטוזומלית דומיננטית עם חדירות 70%, III-4 היא בת בריאה לאם חולה ולאב בריא. מהו הסיכון שהיא נושאת את הגנוטיפ החיובי?
- 50%, כי בכל מחלה דומיננטית בת של חולה נושאת בהכרח 1/2 סיכון בלי קשר לחדירות
- 3/13, כי מעדכנים את ה־Prior (1/2) לפי ההסתברות להיות בריאה אם נושאת (0.3) מול אם אינה נושאת (1)
- 30%, כי זו ההסתברות היחידה להיות בריאה אם נושאת מוטציה (1−0.7)
- 0%, כי אדם בריא לא יכול לשאת גנוטיפ דומיננטי למחלה
פתרון
התשובה הנכונה היא (2).
אנליזה בייסיאנית עם חדירות חלקית:
| נושאת גנוטיפ חיובי | לא נושאת | |
|---|---|---|
| Prior | 1/2 | 1/2 |
| Conditional (בריאה) | 0.3 | 1 |
| Joint | 1/2 × 0.3 = 3/20 | 1/2 × 1 = 1/2 |
| Posterior | (3/20) / (3/20 + 1/2) = 3/13 | 10/13 |
III-4 בריאה, אך בגלל חדירות של 70%, גם אדם עם גנוטיפ חיובי יכול להיות בריא (הסתברות 0.3). לכן הסיכון אינו 0, ואינו 50% - הוא מעודכן ל־3/13 (≈23%).
למה האחרות שגויות:
- (א) החדירות רלוונטית מאוד - היא משמשת כ־Conditional probability.
- (ג) 30% הוא ה־Conditional ולא ה־Posterior.
- (ד) בחדירות חלקית, אדם בריא עדיין עלול לשאת את הגנוטיפ.
מקור: שקפים 74–75
שאלה 22: הבחנה בין דפוסי תורשה בעץ משפחה
אם חולה מעבירה את המחלה לכל ילדיה (בנים ובנות), ואב חולה מעביר את המחלה לכל ילדיו (בנים ובנות). אין דילוג על דורות. מה דפוס ההורשה הסביר?
- אוטוזומלי דומיננטי
- מיטוכונדריאלי
- X-linked dominant
- לא ניתן להבחין ללא בדיקה גנטית
פתרון
התשובה הנכונה היא (1).
המפתח הוא שגם אב חולה מעביר לבנים ולבנות. בתורשה מיטוכונדריאלית, אב חולה לא מעביר כלל (רק אם מעבירה). ב־X-linked dominant, אב חולה מעביר לכל בנותיו אך לאף אחד מבניו. כאן שני ההורים מעבירים לשני המינים ← אוטוזומלי דומיננטי.
סימני היכר להבחנה:
- AD: שני המינים, כל דור, אב מעביר לבנים ולבנות.
- מיטוכונדריאלי: רק אם מעבירה, לכל ילדיה.
- XLD: אב חולה ← כל הבנות חולות, אף בן לא חולה.
למה האחרות שגויות:
- (ב) בתורשה מיטוכונדריאלית אב חולה לא יעביר לילדיו כלל.
- (ג) ב־XLD אב חולה לא יעביר לבנים - כאן הוא כן מעביר.
- (ד) ניתן להבדיל על סמך דפוס ההעברה מהאב.
מקור: שקפים 85–86
שאלה 23: Kearns-Sayre Syndrome
מהי תסמונת Kearns-Sayre וכיצד היא מורשת?
- נגרמת ממוטציית missense בגן גרעיני ועוברת בתורשה אוטוזומלית רצסיבית
- נגרמת ממחיקה גדולה ב־mtDNA, לרוב מופיעה de novo ולא בתורשה אימהית “קלאסית” מאם חולה
- נגרמת ממוטציה ב־mtDNA אך עוברת תמיד בתורשה אימהית עם חדירות מלאה
- נגרמת מכפילות של כל mtDNA וגורמת לעודף ייצור אנרגיה ברקמות
פתרון
התשובה הנכונה היא (2).
תסמונת Kearns-Sayre נגרמת ממחיקה (Deletion) גדולה ב־mtDNA. בדרך כלל היא לא מורשת מאם חולה אלא מופיעה de novo - המחיקה מתרחשת בביצית של אם בריאה, או שאחוז קטן מהביציות נושא את המחיקה (Gonadal mosaicism). בנוסף, מחיקות סומטיות נרכשות של mtDNA יכולות להוביל למחלות ניווניות - למשל בתאים דופאמינרגיים.
למה האחרות שגויות:
- (א) הגורם הוא מחיקה ולא מוטציית Missense, והיא לרוב אינה מורשת קלאסית מאם חולה.
- (ג) KSS נגרמת ממוטציה ב־mtDNA עצמו - לא ב־DNA גרעיני.
- (ד) מחיקה (לא כפילות) ← ירידה בתפקוד OXPHOS, לא עודף אנרגיה.
מקור: שקף 97
שאלה 24: תורשה כפולה - גרעינית ומיטוכונדריאלית
מה המשמעות מבחינת דפוסי תורשה של מחלות OXPHOS?
- כל מחלות OXPHOS עוברות אימהית בלבד כי התהליך מתרחש במיטוכונדריה
- מחלות OXPHOS יכולות לעבור בכל דפוס (AD/AR/X-linked) אם המוטציה בגן גרעיני, או אימהי אם המוטציה ב־mtDNA
- מחלות OXPHOS עוברות תמיד AR כי רוב החלבונים מקודדים בגרעין
- מחלות OXPHOS לא יכולות להיות X-linked כי גני OXPHOS נמצאים רק על אוטוזומים
פתרון
התשובה הנכונה היא (2).
כ־80 חלבונים של קומפלקס OXPHOS מקודדים ע”י DNA גרעיני, ו־13 חלבונים מקודדים ע”י mtDNA. לכן: מוטציה בגן גרעיני יכולה לגרום למחלת OXPHOS עם דפוס תורשה אוטוזומלי דומיננטי, רצסיבי, או X-linked - תלוי באיזה גן מדובר ועל איזה כרומוזום הוא נמצא. מוטציה ב־mtDNA תוביל לדפוס תורשה אימהי.
“A respiratory chain deficiency can give rise to any symptom in any organ or tissue at any age with any mode of inheritance.”
למה האחרות שגויות:
- (א) לא רק תורשה אימהית - מוטציות בגנים גרעיניים שמקודדים לחלבוני OXPHOS יכולות להוביל לדפוסים אחרים.
- (ג) גם תורשה דומיננטית ו־X-linked אפשריות - לא רק רצסיבית.
- (ד) ייתכנו גני OXPHOS גם על כרומוזום X.
מקור: שקפים 102, 104
שאלה 25: אתגרים באבחון טרום-לידתי של מחלות מיטוכונדריאליות
מדוע אבחון טרום-לידתי של מחלות מיטוכונדריאליות מאתגר במיוחד?
- mtDNA אינו ניתן לריצוף ולכן אי אפשר לזהות מוטציות בעובר
- רמת הטרופלסמיה בדגימות (CVS/מי שפיר) לא בהכרח מייצגת רקמות קריטיות; Bottleneck יוצר שונות גדולה; וסיכון החזרה יכול לנוע בטווח רחב מאוד
- כל המוטציות המיטוכונדריאליות הן סומטיות ולכן אינן קיימות בעובר
- בדיקות טרום-לידתיות אסורות כאשר מדובר במחלות מיטוכונדריאליות
פתרון
התשובה הנכונה היא (2).
אבחון טרום-לידתי של מחלות מיטוכונדריאליות מאתגר מכמה סיבות: סיכון החזרה נע בין 0% ל־100% ולא ניתן לחזותו, רמות ההטרופלסמיה בדגימת CVS או מי שפיר עשויות להיות שונות מהרקמות שיושפעו בעתיד (מוח, לב), ותופעת ה־Bottleneck - מעבר דרך מספר מצומצם של מיטוכונדריות בביציות - גורמת לשונות גדולה ברמות ההטרופלסמיה בין ילדים לאותה אם. לכן ייעוץ גנטי מורכב. חלופה אפשרית: שימוש בביצית תורמת.
למה האחרות שגויות:
- (א) mtDNA ניתן לריצוף ללא בעיה - האתגר הוא בפרשנות, לא בטכנולוגיה.
- (ג) מוטציות מיטוכונדריאליות יכולות להיות גם תורשתיות (מהאם) ולא רק סומטיות.
- (ד) אין איסור חוקי - האתגר הוא מדעי וקליני.
מקור: שקפים 105–106