1) רעיון־על: מי “מטיס” את התא?
המרצה תיאר את התא כמו מערכת עם “טייס אוטומטי” שמחזיק הומאוסטזיס: התא קולט את הסביבה ומשנה ביטוי גנים בהתאם. כשהווירוס נכנס, הוא מנסה “להשתלט על ההגה”: הוא מפעיל סיגנלים שמתאימים לו, מדכא סיגנלים שמפריעים לו, ומכוון את התא לשרת את צרכי ההתרבות הוויראלית.
2) מה הווירוס משנה בתא (הצד של הווירוס)
א. סיגנלינג ותא-מחזור
מיד אחרי הדבקה מתרחש cell signaling. הווירוס:
- ידחוף סיגנלים שעוזרים לו (למשל כאלה שמקדמים חלוקה).
- יעכב סיגנלים שלא טובים לו, במיוחד מוות תאי מתוכנן (Apoptosis).
ב. שינוי ביטוי גנים
המרצה הבחין בין שני מצבים:
-
Lytic infection (מחזור ליטי):
הווירוס “מכבה” את התא לטובת עצמו:
- עוצר/מקטין סינתזות תאיות.
- יכול למנוע יציאת mRNA מהגרעין לציטופלזמה.
- יכול לקדם דגרדציה של mRNA.
-
Latency (לטנטיות): כאן הווירוס רוצה להישאר לאורך זמן, ולכן לא מכבה את התא באופן גורף, אלא עושה שינויי ביטוי גנים ממוקדים: חלק עולים וחלק מושתקים, כדי להתאים את התא ל”אירוח” ממושך.
ג. מטבוליזם ותשתיות שעתוק/רפליקציה
הווירוס יכול לשנות:
- צריכת גלוקוז.
- ליפיד מטבוליזם. מטרת השינויים: יותר אנרגיה ומשאבים, יצירת replication centers / replication factories, ייצור ויריונים והרכבה/שחרור בהמשך.
ד. ארגון הגרעין והציטופלזמה
הווירוס מסוגל “לשפץ” את מבנה התא והגרעין כדי לבנות “מפעלים” לרפליקציה.
3) למה הדבקה לא תמיד נראית אותו דבר?
המרצה הדגיש שכל הדבקה “חדשה” לא צפויה, כי היא תלויה ב:
- איזה נגיף הגיע.
- דרך החדירה (עור / עיניים / נשימה וכו’).
- מינון נגיפי (כמה חלקיקים הגיעו).
- מי המאכסן (מבוגר עם מערכת חלשה, ילד עם מערכת לא בשלה).
4) דוגמת מסלול הדבקה: Ectromelia virus בעכבר
המרצה נתן דוגמה למהלך הדבקה מדורג:
- כניסה דרך חתך בכפות הרגליים (פצע).
- רפליקציה מקומית.
- מעבר לבלוטת לימפה קרובה.
- וירמיה ראשונית (דם) ← הגעה לאיברים כמו כבד וטחול.
- רפליקציה באיברים.
- וירמיה שניונית בכמות גבוהה.
- הגעה לעור ← פצעים/כיבים שיכולים לגדול ולהוביל למוות בעכבר.
הוא ציין שזה poxvirus, מאותה “משפחה” כללית בהקשר של smallpox/vaccinia. זה DNA virus גדול שנשאר בציטופלזמה.
5) למה הדבקה יכולה “להיכשל”?
המרצה מנה כמה סיבות אפשריות:
- הגיעו מעט נגיפים, ומתוך כלל החלקיקים רק חלק קטן באמת אינפקטיבי.
- הנגיף נשאר מחוץ למאכסן ולא “הצליח לתפוס”.
- הוא נספח אבל לתאים מתים (אין רפליקציה).
- הוא נקשר לתא לא רגיש.
- הוא נכנס לתא אבל מתפרק בליזוזום.
6) שכבות ההגנה של הגוף/התא (הצד של המאכסן)
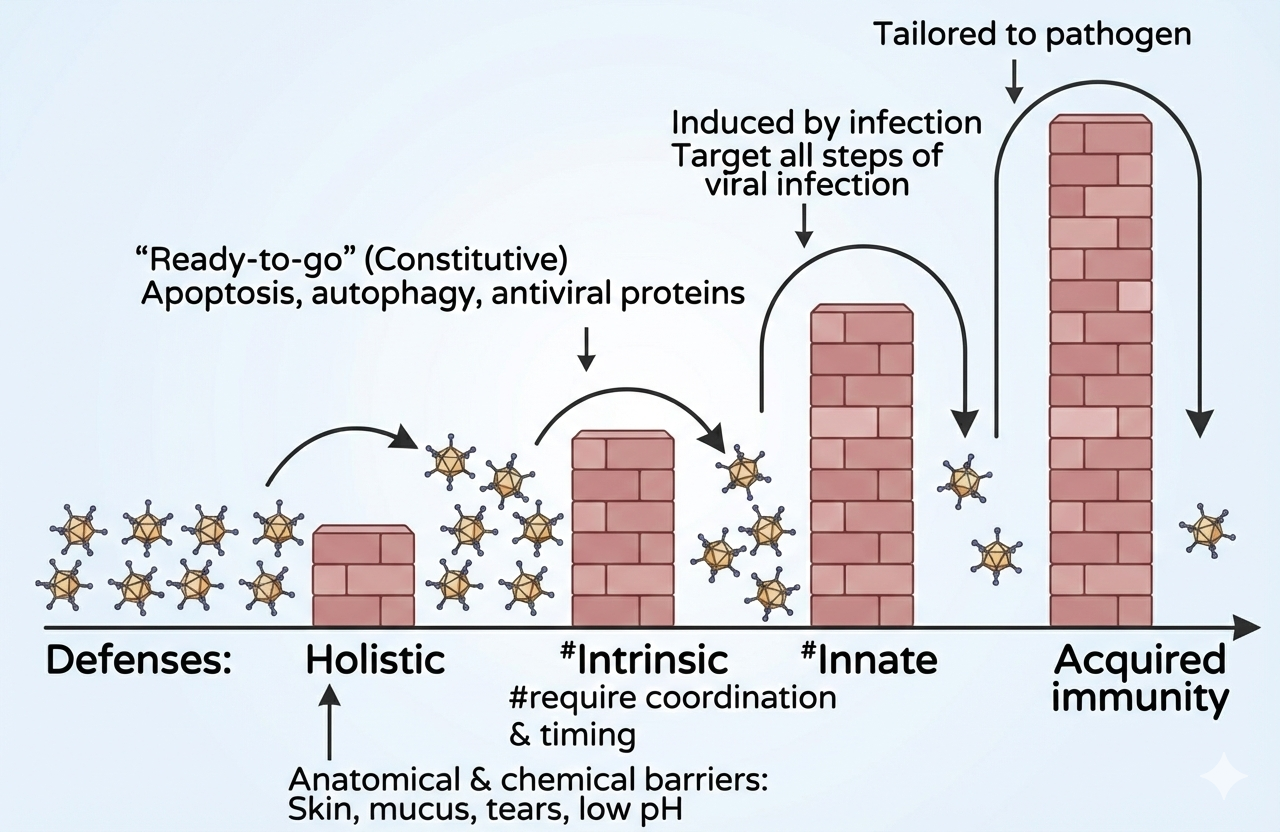
שכבה 1: מחסומים פיזיים-כימיים (Holistic barriers)
- עור כמחסום.
- מוקוס (לוכד פתוגנים; יש בו גם נוגדנים ואנזימים).
- דמעות (הוזכר ליזוזים).
- קיבה: pH נמוך + אנזימים שמפרקים.
שכבה 2: Intrinsic immunity (מוכנה מיד)
המרצה הזכיר מערכות תאיות כמו:
- microRNA / siRNA / CRISPR (הוזכרו כמערכות מורכבות שיילמדו בהמשך).
- Apoptosis
- Autophagy
- אנטי-ויראל פרוטאינס (בהמשך נתן דוגמאות ספציפיות כמו ZAP, PKR ועוד).
שכבה 3: Innate response (דורשת “התחממות”)
זו תגובה של דקות-שעות, כי צריך ביטוי גנים. כאן נכנסים בעיקר:
- Interferons (ציטוקינים שמשרים מצב אנטי-ויראלי בתא ובשכנים).
שכבה 4: Adaptive immunity (איטית יותר)
מעורבים תאי B ו־T ונוגדנים, אך זה לוקח שבועות עד שנבנית תגובה מלאה.
7) דוגמה שמדגישה “דרך הדבקה משנה הכול”: Vaccinia בארנבות
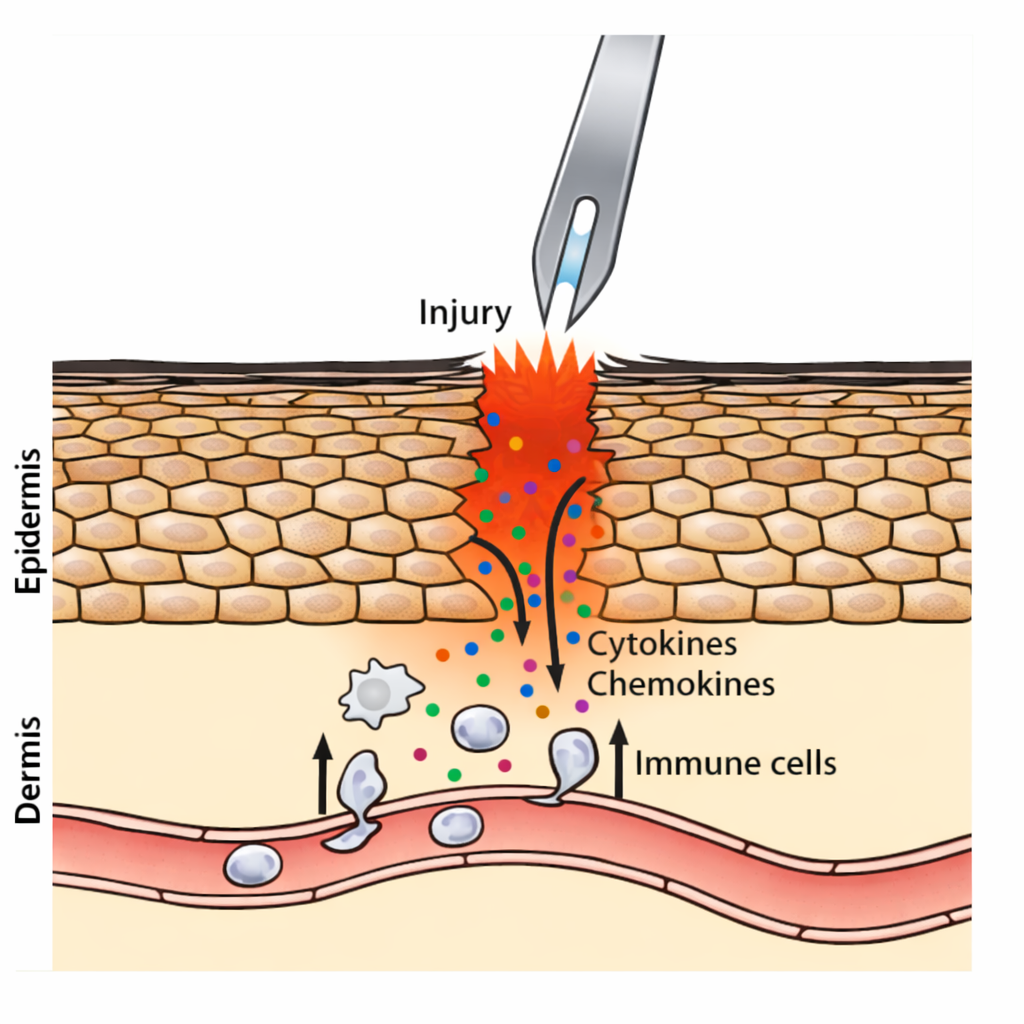
השוואה בין שתי דרכי חשיפה לאותו נגיף:
- הזרקה תת־עורית ← אחרי 8~ ימים כל הארנבות מתות.
- כניסה דרך פציעה בעור ← נוצרת דלקת מקומית, קרישה וסגירת כלי דם, תאים בולעניים מגיעים, מציגים אנטיגן בלימפה ← הארנבות לא מתות ואף מפתחות חיסון להמשך.
המסר: פצע יוצר גם בידוד של ההתפשטות וגם הפעלה יעילה של מערכת החיסון.
8) ספציפיות של שפעת: למה בעיקר בדרכי הנשימה העליונות?
המרצה חזר על מנגנון סביב חלבון המעטפת HA:
- כשהנגיף יוצא מהתא, HA הוא במצב לא פעיל (הוזכר כ־HA0).
- כדי להפוך פעיל, צריך חיתוך ע”י אנזים.
- רק בתאים מסוימים בדרכי הנשימה העליונות (Club cells) יש הפרשה של האנזים הזה.
לכן השפעת לרוב נשארת ממוקדת שם ולא מתפשטת לכל הגוף.
המרצה גם תיאר מצב מסוכן:
- מוטציה בשפעת שהכניסה שיירים בסיסיים כך שהחיתוך מתבצע ע”י אנזים נפוץ בהרבה תאים ← הנגיף נמצא גם בטחול/כבד/ריאות/כליות/מוח. הוא תיאר אירוע בלול תרנגולות שהוביל להשמדה מחשש להדבקה בבני אדם.
9) איך התא “מבין” שיש הדבקה ומדליק אזעקה?
המרצה נתן שתי דרכים עיקריות:
א. “משהו לא לפי התוכנית”
שינויים ב:
- מטבוליזם
- סינתזת חלבון
- cell cycle
התא “מרגיש” שזה לא הרוטינה התקינה.
ב. חיישנים שמזהים “לא־עצמי” (Pattern Recognition Receptors)
נקודות חשובות:
- TLRs: על ממברנת התא או בתוך אנדוזום (רלוונטי כי הרבה וירוסים נכנסים באנדוציטוזיס). הדגש היה לא על לזכור מספרים, אלא להבין שיש חישה של חלבונים ונוקלאיק אסידס ויראליים.
- RIG-I (ציטופלזמי): מזהה למשל dsRNA או RNA בלי cap/עם קצה 5’ פוספט.
- cGAS-STING: DNA בציטופלזמה מפעיל cGAS ← יצירת שליח שניוני (cGAMP) ← STING נע ל־Golgi ומפעיל מסלולי תגובה.
- הוזכרו גם חיישנים נוספים (כולל כאלה יותר “חיידקים/פטריות” בהקשר CLR).
מה יוצא מהחישה? המסר הקריטי: זה מוביל להפעלת טרנסקריפשן פקטורס (הוזכרו IRF3/IRF7 וגם NF-κB) ← ביטוי אינטרפרונים.
10) אינטרפרון (Interferon): איך זה עובד ולמה זה גם “עונש” לתא?
מה זה אינטרפרון? (IFN)
קבוצת ציטוקינים שמופרשים מתאים מודבקים (וגם ממערכת החיסון), נקלטים באותו תא ובתאים סמוכים, ומפעילים כמעט 1000~ גנים (ISGs - “interferon-stimulated genes”).
אי אפשר להשאיר את תגובת האינטרפרון פעילה כל הזמן (ובמיוחד את ביטוי ה־ISGs), כי היא מעכבת תהליכים חיוניים בתא ועלולה לגרום לנזק תפקודי ואף למוות תאי.
הניסוי ההיסטורי שהוביל לגילוי האינטרפרון
תצפית משנות ה־50:
- מדביקים תאים בווירוס שעבר UV inactivation (לא יכול להתרבות אבל עדיין “נראה” לתא).
- לוקחים את המדיום שבו התאים היו (רק מה שהתאים הפרישו).
- מעבירים את המדיום לתאים אחרים, נותנים זמן (כי צריך ביטוי גנים), ואז מדביקים.
- התאים החדשים מוגנים ← היה “משהו שמפריע להדבקה” ← Interferon.
סוגי אינטרפרונים
- Type I: IFN-α (בעיקר מתאים של מערכת החיסון כמו דנדריטיים), IFN-β (יכול להיות מהרבה תאים מודבקים).
- Type II: IFN-γ (מופרש ע”י NK ותאי T מופעלים, וגם בהקשר למקרופגים).
- Type III: הוזכר שיש גם קבוצה כזו (בלי להיכנס לפרטים).
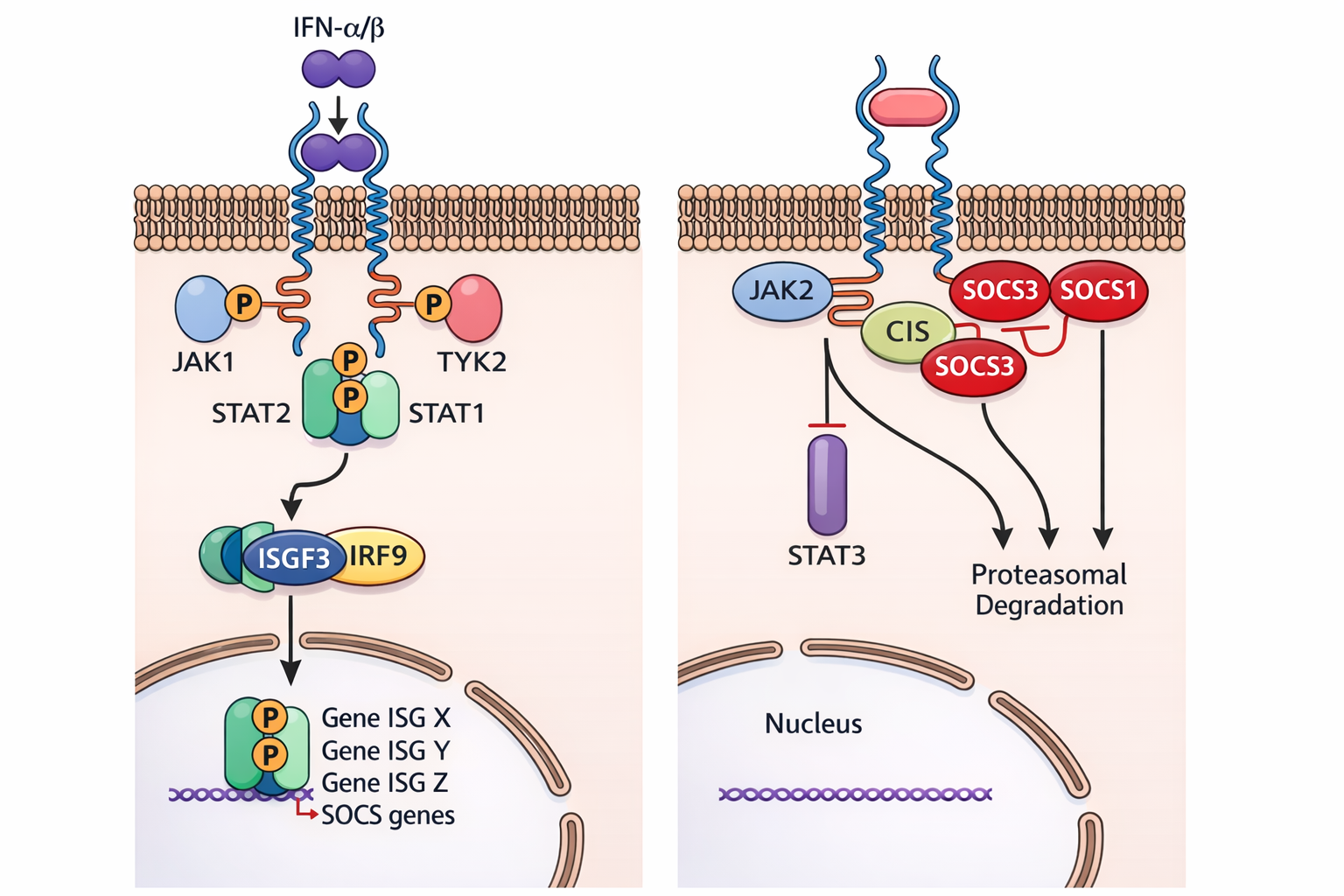
מסלול האיתות (העיקר)
קישור לרצפטור ← דימריזציה ← JAK1 ← פוספורילציה של STAT ← כניסה לגרעין ← הפעלת ISGs.
מטפורה: “כיבוי שריפה”
המרצה תיאר את זה כמו ליצור “אזור חיץ” סביב מוקד ההדבקה: התאים מסביב נכנסים למצב אנטי-ויראלי כדי לעצור התפשטות.
למה זה גורם לנו להרגיש חולים?
אינטרפרונים/ציטוקינים גורמים:
- חום, צמרמורות, הרגשה רעה (“flu-like symptoms”).
דגש: לא הנגיף עצמו “עושה” את התחושה, אלא תגובת הגוף.
בעבר, היו טיפולים באינטרפרון חודשים ארוכים עם סבל משמעותי.
איך הגוף “מוריד את הווליום” אחרי שהכול נגמר?
הוזכר גן כמו SOCS (interferon-responsive) שתפקידו לעכב את סיגנל האינטרפרון כדי לחזור לשגרה.
11) דוגמאות לכלים אנטי-ויראליים שהאינטרפרון מעלה (ISGs)
המרצה נתן קשת של “נקודות עצירה”:
- חסימת כניסה או חסימת כניסת הגנום לגרעין.
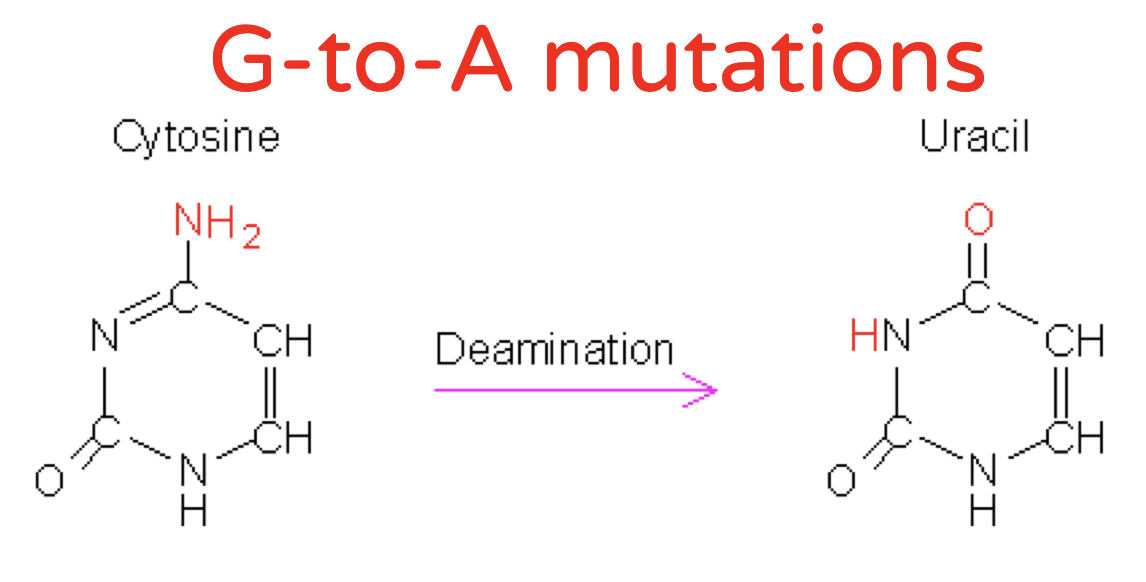
- יצירת מוטציות בווירוס: APOBEC.
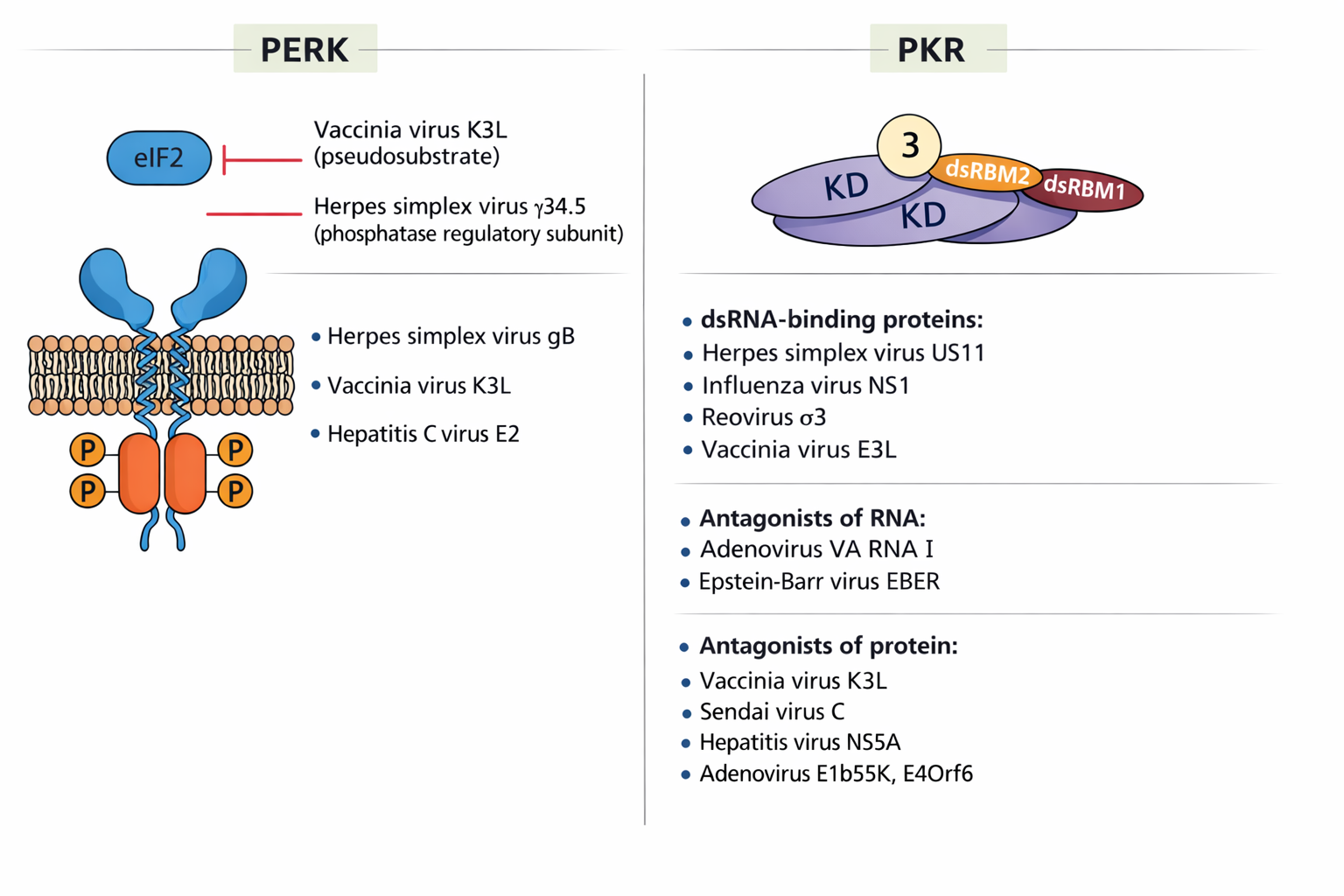
- עיכוב תרגום: PKR ← פוספורילציה של eIF2 ← עצירת cap-dependent translation.
- דגרדציה של RNA חריג: ZAP (זיהוי CpG גבוה ב־RNA).
- עיכוב assembly ושחרור: Tetherin (דוגמה מרכזית).
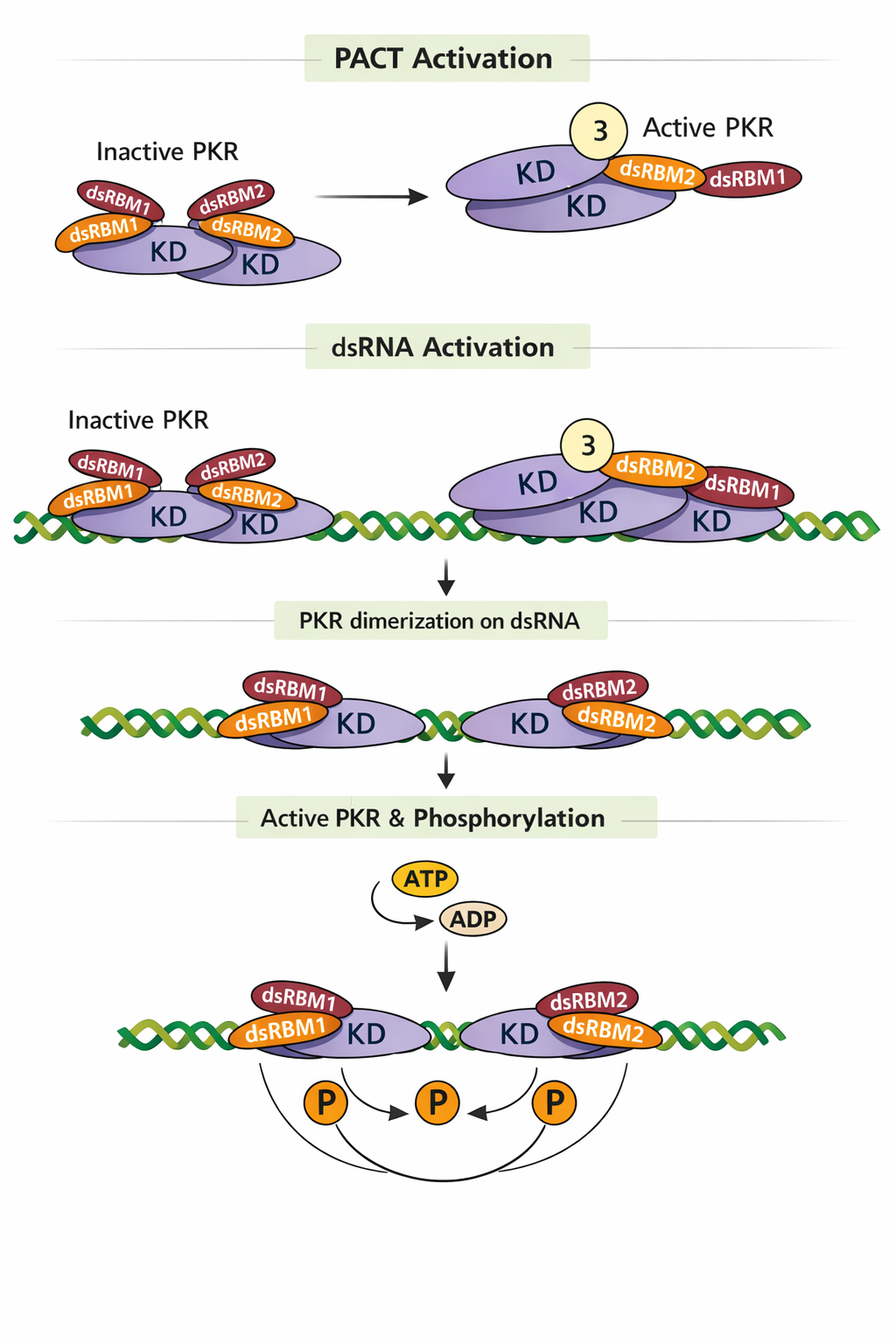
- זיהוי החלבון: האיור מתמקד בחלבון PKR (שמופיע בכותרות ובתוך השרטוט).
- המנגנון: האיור מראה כיצד PKR, שהוא בדרך כלל לא פעיל (Inactive), מזהה dsRNA (רנ”א דו־גדילי, סימן מובהק לנוכחות נגיפית) או מופעל ע”י חלבון עקה (PACT).
- התוצאה: האיור מראה שבסוף התהליך ה־PKR עובר זרחון (הוספת P, או Autophosphorylation). זהו בדיוק השלב ש”דורך את הנשק” ומאפשר ל־PKR לבצע את מה שכתוב בסיכום: לעצור את ייצור החלבונים בתא כדי למנוע מהווירוס להתרבות.
Tetherin + דוגמה עם HIV
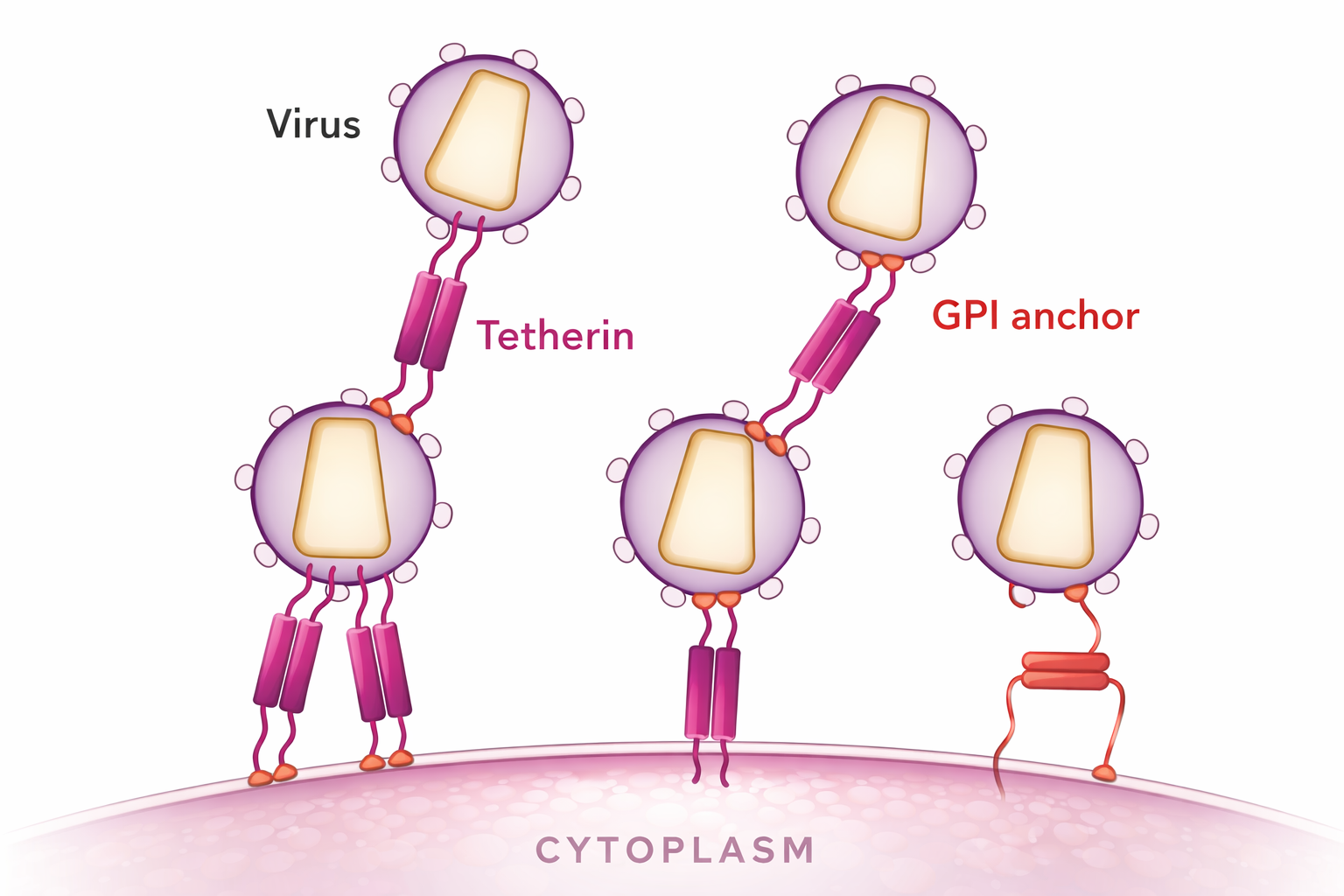
Tetherin “קושר” את הוויריון לממברנת התא ומונע שחרור. המרצה תיאר ניסויים שבהם:
- בלי Vpu (חלבון HIV שמעכב tetherin) ← הרבה ויריונים “תקועים” על הממברנה.
- גם כשהעלו מאוד tetherin, הווירוס לא תמיד מצליח לעכב את כולו.
מסר: יש “משיכת חבל” אבולוציונית - התא מפתח חסימה והווירוס מפתח אנטגוניסטים.
דוגמת APOBEC מול HIV
החלבון APOBEC (APOBEC3G) עושה דה־אמינציה של ציטוזין וגורם להמון מוטציות, כך שנוצר וירוס לא אינפקטיבי.
HIV משתמש ב־Vif:
- שולח APOBEC לדגרדציה באמצעות יוביקוויטין.
- וגם יכול למנוע ממנו להיכנס לקפסיד.
12) עוד מנגנונים תאיים שהוזכרו
Apoptosis
המרצה הסביר שזה “לטובת הגוף השלם”: התא מתפרק בצורה מבוקרת ונבלע ע”י מקרופגים. הוא הדגיש שווירוסים רבים מעכבים אפופטוזיס, אבל לפעמים דווקא מפעילים אותו בשלב מאוחר - ייתכן כדי לעזור להפצה (הועלו רעיונות בכיתה).
Autophagy
התא יכול “לעטוף” מרכיבים בעייתיים (כולל וירוסים) בממברנה, להתאחות עם ליזוזום ולפרק.
וירוסים (למשל הרפסים) יכולים לקודד הרבה חלבונים שמעכבים אוטופאג’י.
השתקת DNA ויראלי ככרומטין
הוזכר: התא יכול “לארוז” DNA ויראלי ולהפוך אותו להטרוכרומטין כדי להשתיק. קומפלקסים כמו PML bodies מעורבים, ורמת PML עולה בתגובה לאינטרפרון.
דוגמה: ב־EBV (הרפס) יש חלבונים שיכולים לפרק/לשנות את PML כדי לאפשר מעבר לליטי, בעוד שבלטנטיות הווירוס יכול “ליהנות” מהשתקה.
13) וירוסים לא נשארים חייבים: אנטגוניזם למסלולים
לכל מסלול הגנה תאית (RIG-I, MAVS, cGAS-STING, IRF/NF-κB, tetherin וכו’) וירוסים רבים מקודדים חלבוני עיכוב:
- קישור שמונע הפעלה,
- דה־יוביקוויטינציה שמונעת סיגנלינג,
- דגרדציה של רכיבי המסלול,
- או “הומולוגים” ויראליים שמתחרים על DNA בלי יכולת להפעיל שעתוק.
לכאורה, לא צריך לזכור שמות (מניסיון - קחו זאת בערבון מוגבל) - כן צריך להבין את עקרון “שחמט” של מהלכים ומהלכי-נגד.
14) שונות גנטית במאכסן ורזרבוארים
- בזני עכברים שונים יש תגובה שונה לאינפלואנזה, בגלל הבדלים (כמו SNPs) ברצפטורים/מסלולים (TLR וכד’).
- גם באוכלוסייה האנושית יש וריאנטים (הוזכרו IFITM, TLR3) שיכולים להעלות רגישות לשפעת.
- הטלפים הוצגו כ”אלופים”: חיים שנים רבות, חיים בצפיפות, ויש אצלם מערכת אינטרפרון פעילה מאוד באופן קבוע, ולכן הם יכולים לשמש רזרבואר לנגיפים בלי לחלות, אך להעביר לאחרים.
15) המסר המסכם של השיעור
מערכת האינטרפרון היא נשק חזק נגד וירוסים, אבל אם לא מווסתים אותה נכון, היא יכולה להזיק ואף לתרום למצבים פתולוגיים (המרצה הזכיר גם אוטואימוניות כעיקרון כללי של “תגובה לא מבוקרת”).
שאלה לדוגמה: על אילו תאים משפיע האינטרפרון?
התשובה: גם על התאים המודבקים וגם על התאים הסמוכים.
נקודות חשובות
- לא לזכור מספרי TLR/רשימות ארוכות של חלבונים ויראליים מעכבים.
- כן להבין: שכבות ההגנה, לוגיקת החישה, אינטרפרון ← JAK/STAT ← ISGs, והרעיון של מרוץ חימוש (host vs virus).