פתיחה - למה מתחילים מספריות DNA?
בחלק הבא של הקורס נעסוק בעיקר ב־DNA damage וב־Genome instability - נזקי DNA ואי־יציבות גנומית. נלמד אילו סוגי נזק יכולים להתרחש, מה גורם להם, איך התא מתמודד איתם, ומהן ההשלכות האפשריות שלהם: שינויים כרומוזומליים, סרטן, הזדקנות ופתולוגיות נוספות.
אבל לפני שנוכל לדבר על זיהוי ומיפוי של נזקי DNA, צריך להבין את הכלים שבאמצעותם עושים זאת במעבדה. חלק גדול מהזיהוי של נזקי DNA, של שינויים כרומוזומליים ושל מצבים אפיגנטיים נעשה היום בעזרת ריצוף גנומי. לכן שני השיעורים הראשונים בפרק הזה עוסקים בספריות DNA.
מהי ספריית DNA?
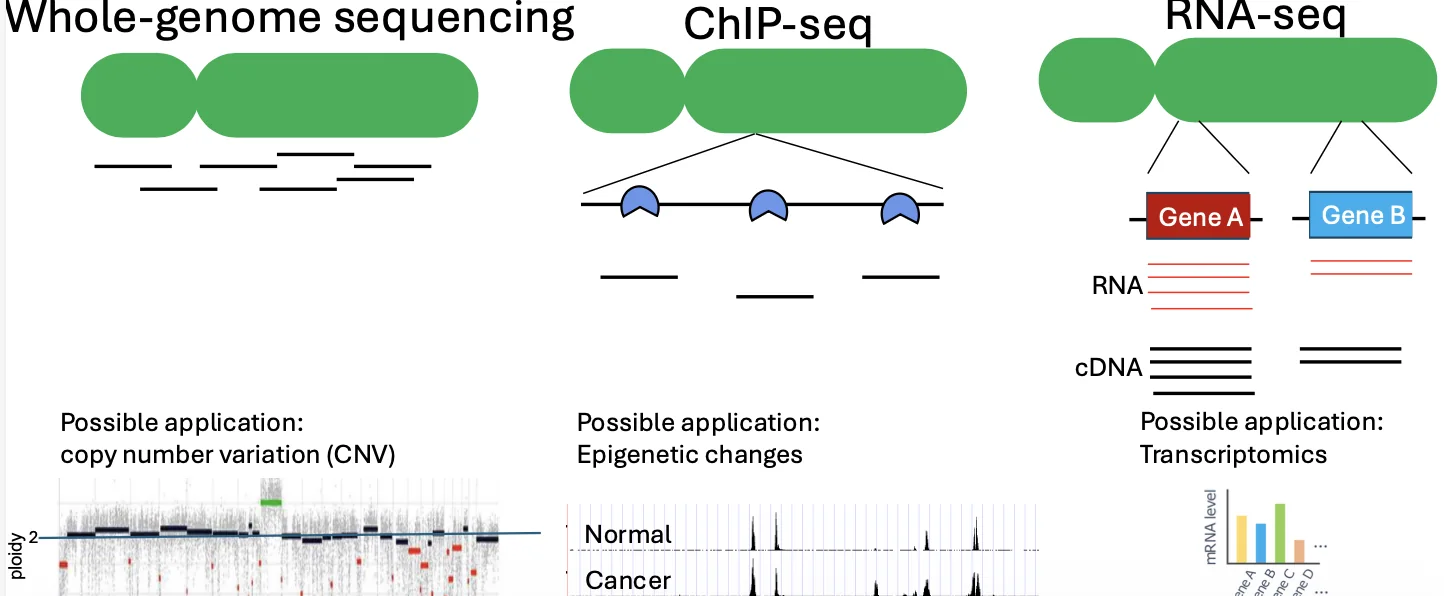
ספריית DNA היא אוסף מקטעי DNA שהוכנו כך שאפשר יהיה לרצף אותם.
השאלה החשובה היא: אילו מקטעים נכנסים לספרייה? התשובה תלויה במטרת הניסוי.
לדוגמה:
- אם המטרה היא לרצף את כל הגנום, הספרייה תכיל מקטעים מכל רחבי הגנום.
- אם המטרה היא לבדוק איפה חלבון מסוים נקשר ל־DNA, הספרייה תכיל בעיקר מקטעים מאותם אזורים שאליהם החלבון נקשר.
- אם המטרה היא לבדוק ביטוי גנים, המקור הראשוני יהיה RNA ולא DNA, אבל במהלך הכנת הספרייה ממירים אותו ל־cDNA, כלומר ל־DNA משלים, ורק אז נרצף אותו.
הרעיון המרכזי: ספרייה היא אוכלוסיית המקטעים שנבחרו והוכנו לריצוף, לפי השאלה הביולוגית הנשאלת.
סוגים מרכזיים של ספריות DNA
| סוג הספרייה | מקור הדאטה | מה בודקים? | שימוש עיקרי |
|---|---|---|---|
| Whole Genome Sequencing גנומי · כל הגנום | DNA גנומי | כל הגנום | מוטציות, מחיקות, אמפליפיקציות ו־Copy Number Variation |
| Whole Exome Sequencing גנומי · אזורים מקודדים | DNA גנומי | אקסונים מתוך ה־DNA | מוטציות ושינויים באזורים מקודדים |
| RNA-seq טרנסקריפטומי | RNA שעובר המרה ל־cDNA | רמות ביטוי של גנים | אנליזה טרנסקריפטומית, ולעיתים גם Alternative splicing |
| ChIP-seq קישור חלבונים / אפיגנטיקה | DNA הקשור לחלבון/מודיפיקציית היסטון | מיקומי קישור בגנום | מיפוי פקטורי שעתוק, היסטונים ומודיפיקציות אפיגנטיות |
| ATAC-seq נגישות כרומטין | DNA מאזורים נגישים בכרומטין | כרומטין פתוח/סגור | מיפוי נגישות כרומטין |
| Methylation-seq מתילציה | DNA גנומי | דפוסי מתילציה | רגולציה אפיגנטית |
| Hi-C ארגון תלת־ממדי | DNA שהיה קרוב במרחב | מבנה תלת־ממדי של הגנום | ארגון הכרומטין במרחב הגרעיני |
Whole exome sequencing לעומת RNA-seq
בשני המקרים ניתן לקבל מידע שממופה בעיקר לאקסונים, אבל מקור המידע שונה:
-
Whole exome sequencing: מרצפים אקסונים מתוך ה־DNA הגנומי. המטרה היא לזהות מוטציות, מחיקות, אמפליפיקציות או שינויים גנומיים אחרים באזורים המקודדים.
-
RNA-seq: מתחילים מ־RNA, הופכים אותו ל־cDNA, ואז מרצפים. לכן המידע משקף בעיקר את מה שבאמת בוטא בתא או ברקמה.
ב־RNA-seq לא כל אקסון חייב להופיע בכל טרנסקריפט, בגלל Alternative splicing. לכן מעבר לרמות ביטוי, בתנאים מסוימים ניתן לזהות גם וריאציות של שחבור.
RNA-seq - אילו גנים פעילים ובאיזו מידה הם משועתקים.
חשיבות ספריות DNA לרפואה
ספריות DNA ו־NGS הן חלק מהתרומות הגדולות של הביולוגיה המולקולרית לרפואה המודרנית. השימושים הקליניים הולכים וגדלים, במיוחד באונקולוגיה, באבחון מולקולרי ובזיהוי פתוגנים.
1. פרופיל גנטי של גידולים - FoundationOne כדוגמה
אחת הדוגמאות היא בדיקות מסוג FoundationOne, שמטרתן לזהות שינויים גנומיים בגידול שיכולים לעזור בבחירת טיפול.
במקום לרצף את כל הגנום, משתמשים בפאנל ממוקד של גנים הקשורים לסרטן, לרוב מדובר בפאנל של מאות גנים, למשל 324 גנים. כך ניתן לבדוק מוטציות, מחיקות, אמפליפיקציות ושינויים נוספים בצורה ממוקדת וזולה יותר.
יש כאן פשרה בין שני עקרונות:
- מצד אחד, הבדיקה לא מכסה את כל כלל השינויים הגנומיים האפשריים בסרטן.
- מצד שני, היא מתמקדת בשינויים שיש להם משמעות טיפולית, כלומר שינויים שהם druggable או רלוונטיים לבחירת טיפול.
EGFR כדוגמה
לדוגמה, שינוי מסוים ב־EGFR יכול לגרום לרצפטור להיות פעיל גם בלי ליגנד. כלומר, האקטיבציה שלו הופכת ל־ligand-independent. מצב כזה יוצר פעילות יתר של המסלול, ולכן יש לו פוטנציאל סרטני.
החשיבות הקלינית היא שאם מזהים את המוטציה הזאת, ניתן להשתמש ב־EGFR inhibitors שמתאימים לגידולים עם השינוי הספציפי הזה.
נקודת המפתח כאן היא לא לזכור חברה מסחרית, אלא להבין את העיקרון: ריצוף ממוקד מאפשר לזהות שינוי מולקולרי בגידול, והשינוי יכול לכוון את בחירת הטיפול.
2. זיהוי פתוגנים מדם
הוזכרה גם בדיקה לזיהוי זיהומים מדגימות דם, כמו KariusTest. הרעיון הוא לרצף DNA מתוך הדוגמה ולהשוות אותו לפתוגנים אפשריים. העיקרון: ריצוף מאפשר לזהות מקור זיהומי לפי חתימות DNA בדגימה. השיעור לא נכנס לפרטים.
3. ניבוי פרוגנוזה לפי RNA-seq
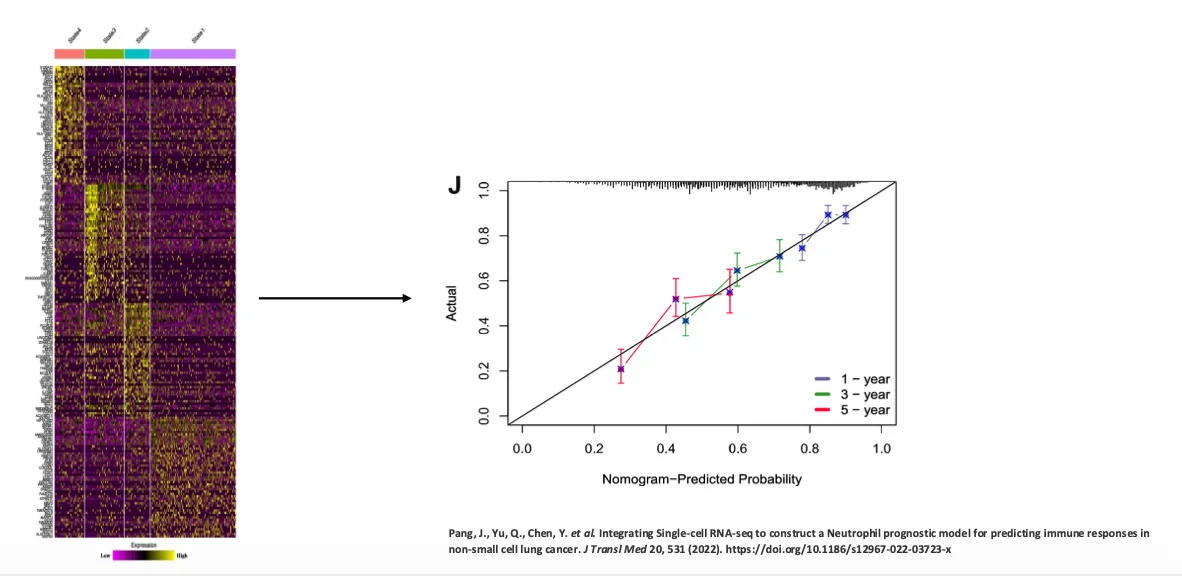
דוגמה נוספת היא שימוש בפרופיל ביטוי גנים כדי לנבא פרוגנוזה בחולי סרטן. לדוגמה, Non-small cell lung cancer.
העיקרון הוא לקחת דגימות RNA-seq מחולים, למדוד את רמות הביטוי של גנים רבים, ולבנות מודל שמנבא הישרדות או פרוגנוזה. זה מקרה של Supervised learning:
- יש לנו נתוני RNA-seq מחולים.
- יש לנו גם מידע ידוע על הפרוגנוזה שלהם.
- המודל לומד את הקשר בין פרופיל הביטוי לבין התוצאה הקלינית.
- אחר כך ניתן להשתמש בו כדי לנבא פרוגנוזה של חולים חדשים.
במודלים כאלה השאלה הראשונית היא לא בהכרח סיבתיות, אלא יכולת ניבוי. אם המודל מנבא טוב, הוא שימושי. לאחר מכן אפשר לבצע הנדסה לאחור ולשאול למה גן מסוים קשור לפרוגנוזה טובה או רעה, אבל זו כבר שאלה אחרת.
פרויקט הגנום האנושי ומהפכת ה־NGS
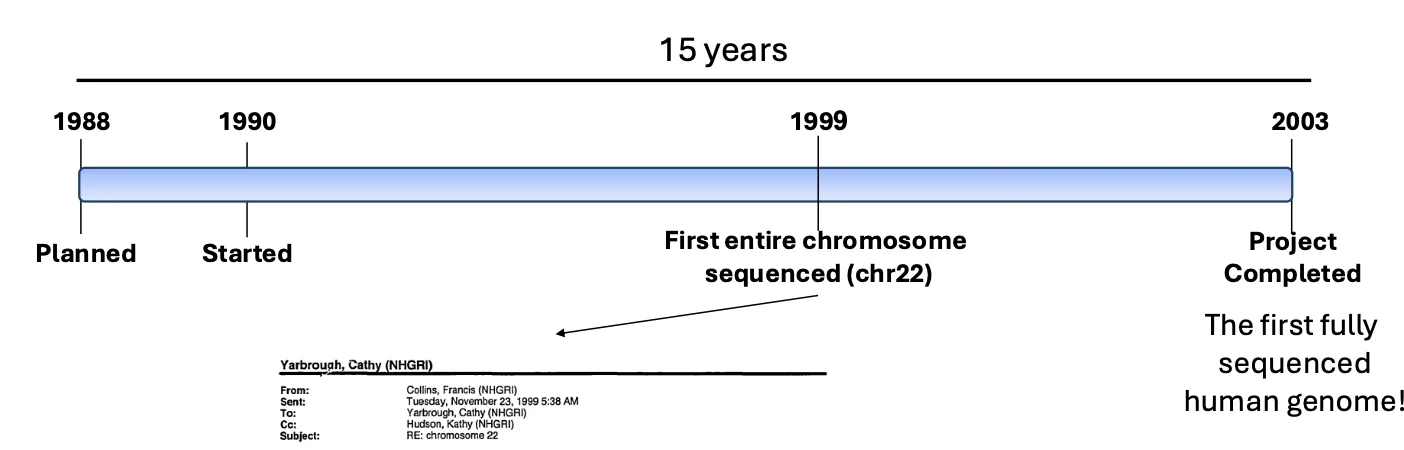
לפני שנכנס ל־NGS, נחזור להיסטוריה של ריצוף הגנום האנושי.
פרויקט הגנום האנושי התחיל רשמית ב־1990, לאחר תכנון שהחל עוד קודם לכן. בשנת 1999 רוצף לראשונה כרומוזום אנושי שלם - כרומוזום 22. בשנת 2003 הושלם הריצוף של הגנום האנושי הראשון.
היקף הפרויקט היה עצום:
- כ־15 שנה של עבודה
- כ־1,000 מדענים
- עלות של כ־3 מיליארד דולר
לעומת זאת, כיום ניתן לרצף גנום שלם בזמן קצר בהרבה, בעלות של פחות מאלף דולר, ובתהליך שיכול להתבצע על ידי סטודנט אחד שלא בהכרח מנוסה. מכאן שהמעבר ל־NGS לא היה התפתחות הדרגתית, אלא מהפכה של ממש.
המהפכה של NGS: המעבר מריצוף איטי, יקר וממוקד לריצוף מקבילי של מיליוני מקטעים, שמאפשר עבודה גנומית בקנה מידה עצום.
Sanger sequencing - דור ראשון
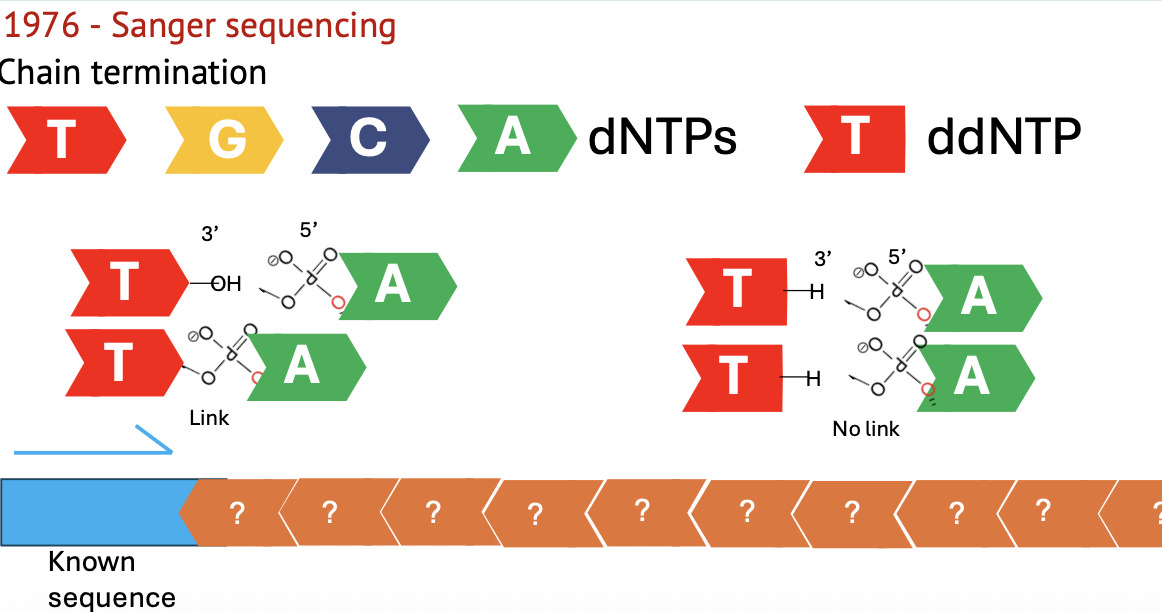
לפני Next Generation Sequencing היה First Generation Sequencing, כלומר Sanger sequencing.
עקרון השיטה
אם יש רצף DNA שאיננו יודעים מהו, אבל יש לנו לפניו רצף ידוע שמאפשר לתכנן primer, ניתן לסנתז את הגדיל החדש במבחנה. בתגובה מכניסים:
- ארבעה dNTPs רגילים
- וגם ddNTP אחד או יותר
ההבדל הקריטי הוא של־ddNTP חסרה קבוצת ה־OH בעמדה 3’. לכן, ברגע ש־ddNTP נכנס לגדיל המסונתז, אי אפשר להוסיף אחריו נוקלאוטיד נוסף והסינתזה נעצרת.
5'
(P)─(P)─(P)─OCH₂ O Base
│ ╱ ╲ │
│╱ H H ╲│
│▚ │ │ ▞│
│ ▚├▄▄▄▄▄▄▄▄▄▄┤▞ │
H │ │ H
[H] H
Dideoxynucleotide (ddNTP)
(P)─(P)─(P)─OCH₂ O Base
│ ╱ ╲ │
│╱ H H ╲│
│▚ │ │ ▞│
│ ▚├▄▄▄▄▄▄▄▄▄▄┤▞ │
H │ │ H
OH H
DNA
אם למשל משתמשים ב־ddTTP, מקבלים תוצרים שמסתיימים בכל מקום שבו היה צריך להיכנס T. לפי אורכי התוצרים אפשר להסיק באילו מיקומים הופיע T. כשעושים את זה לכל ארבעת הנוקלאוטידים, ניתן לשחזר את הרצף.
קריאת התוצרים
בגרסה המקורית, הפרדת התוצרים נעשתה בג’ל לפי גודל. כיום, בסנגר מודרני, משתמשים בכל ה־ddNTPs יחד, אבל כל אחד מסומן ב־fluorophore אחר. התוצאה נקראת כ־chromatogram.
חסרונות Sanger
לשיטת Sanger יש כמה מגבלות חשובות:
- קשה לפתור אזורים עם רצפים חוזרניים.
- קשה לזהות במדויק רצפים של אותו נוקלאוטיד שחוזר הרבה פעמים ברצף.
- התהליך איטי ומוגבל בהיקפו.
- הוא מתאים לרצפים קצרים יחסית, למשל ריצוף פלסמיד או מקטע DNA קטן.
נקודת המעבר ל־NGS: סנגר מצוין כשיש מעט DNA ורצף קצר יחסית. NGS נדרש כשרוצים לרצף הרבה מאוד מקטעים במקביל.
NGS - הרעיון המרכזי
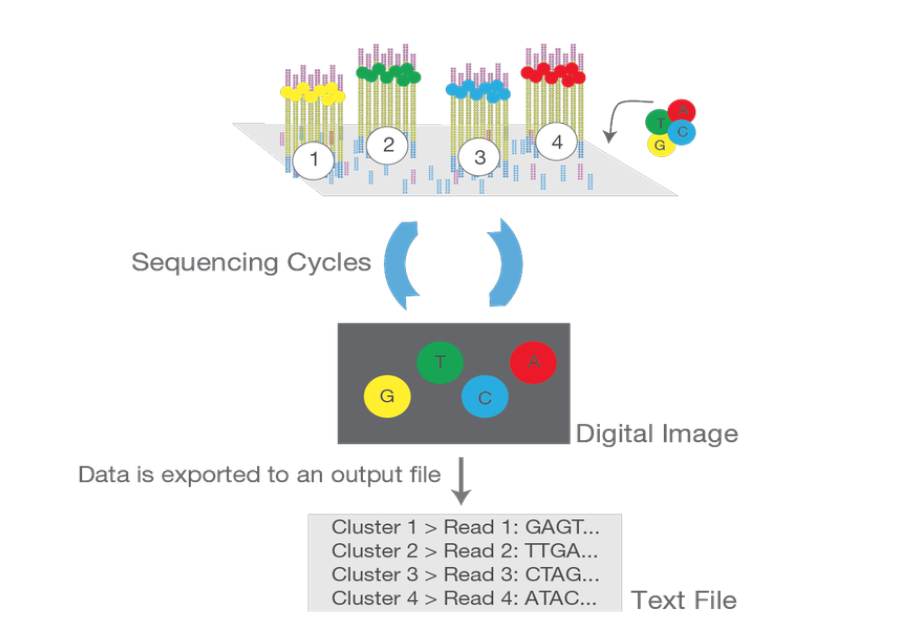
כאשר מדברים על ספריות DNA ו־NGS, אפשר לחלק את התהליך לשלושה חלקים:
- הכנת הספרייה במעבדה - בחירת המקטעים, חיתוך, חיבור אדפטורים, Size selection ולעיתים אמפליפיקציה.
- הריצוף במכונה - קיבוע המקטעים ל־Flowcell, הגברה וריצוף מקבילי.
- אנליזה חישובית - תלויה במטרת הניסוי: Variant calling, Peak calling, Transcriptomic analysis ועוד.
מה הופך את NGS ליעיל?
יש שני עקרונות מרכזיים:
1. Parallelization - ריצוף מקבילי
במקום לרצף מולקולה אחת בכל פעם, מחלקים את הגנום להרבה מקטעים קטנים, ומרצפים את כולם במקביל.
אפשר לחשוב על זה כעל ״הפרד ומשול״:
- חותכים את ה־DNA למקטעים קצרים.
- מחברים להם אדפטורים.
- מקבעים אותם על משטח הריצוף.
- מרצפים מיליוני מקטעים במקביל.
2. Sequencing by synthesis
ב־Sanger קודם מייצרים תוצרים ואז קוראים אותם. ב־NGS, הקריאה מתרחשת בזמן הסינתזה עצמה.
בכל מחזור מתווסף נוקלאוטיד פלואורסצנטי. לכל נוקלאוטיד יש סיגנל פלואורסצנטי אחר, והדטקטור מזהה איזה בסיס נוסף בכל קלאסטר. לאחר מכן ממשיכים למחזור הבא, וכך קוראים את הרצף בסיס אחר בסיס.
Flowcell וקלאסטרים
המקטעים עם האדפטורים נקשרים ל־Flowcell, שהוא משטח שעליו יש רצפים שמזהים את האדפטורים. לאחר הקיבוע, כל מקטע עובר הגברה מקומית, כך שנוצר cluster - הרבה עותקים של אותו מקטע באותו מיקום.
המיקום של הקלאסטר נשמר לאורך הריצוף. לכן גם אם מקטעים שונים אינם בדיוק באותו אורך, זה לא מפריע: כל קלאסטר נקרא בנפרד, בסיס אחר בסיס. כאשר מקטע מסוים נגמר, הוא פשוט מפסיק לתת סיגנל.
הכנת ספריית DNA
הכנת ספרייה משתנה לפי סוג הספרייה, אבל ניתן לחלק אותה לשתי פאזות כלליות:
- פאזה 1 - סלקציה למקטעים שמעניינים אותנו - בשלב זה בוחרים את המקטעים לפי מטרת הניסוי.
- פאזה 2 - שלבים משותפים לרוב הספריות - בשלב זה מבצעים את אותם שלבים טכניים, ללא קשר לסוג הספרייה.
פאזה 1 - סלקציה למקטעים שמעניינים אותנו
זו הפאזה שבה הספריות שונות זו מזו.
- ב־Whole genome sequencing אין סלקציה ביולוגית מיוחדת - לוקחים את כל הגנום.
- ב־ChIP-seq דגים מקטעים שקשורים לחלבון מסוים בעזרת נוגדן.
- ב־RNA-seq מתחילים מ־RNA, הופכים אותו ל־cDNA, ואז ממשיכים להכנת ספרייה.
פאזה 2 - שלבים משותפים לרוב הספריות
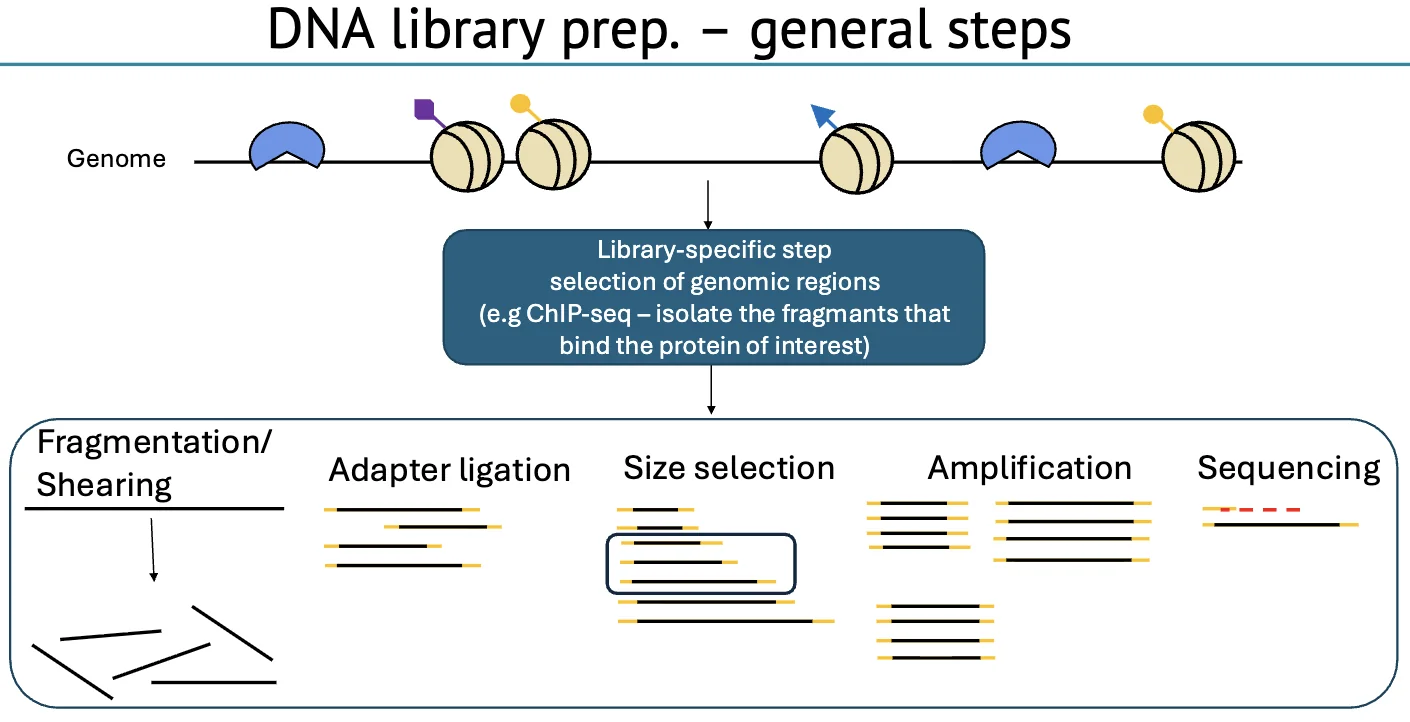
ברוב הספריות חוזרים אותם שלבים טכניים:
- Fragmentation/Shearing - חיתוך ה־DNA למקטעים
- End repair - תיקון הקצוות כך שיהיו נקיים וישרים
- A-tailing - הוספת A בקצה
- Adapter ligation - חיבור אדפטורים עם T-overhang
- Size selection - בחירת מקטעים בטווח גודל מתאים
- PCR amplification - הגברה, אם נדרש
- Sequencing - ריצוף במכונה
פרגמנטציה - איך שוברים DNA?
הדרך הנפוצה היא סוניקציה; מכשיר כמו Covaris משתמש בגלי קול בתווך נוזלי. גלי הקול יוצרים בועות, והבועות מתכווצות ומתפוצצות. הפיצוץ יוצר shear force, כוח מכני שמסוגל לשבור את ה־DNA.
החיתוך נראה אקראי, אבל ניתן לשלוט בפרמטרים של גלי הקול וכך לשלוט בטווח האורכים הרצוי. למשל, ניתן לכוון לקבלת מקטעים בטווח של כ־200–300 בסיסים.
ניקוי הקצוות (End repair)
אחרי סוניקציה מתקבלים קצוות לא אחידים: לפעמים יש overhang חד־גדילי, לפעמים חסר קטע בקצה. כדי לחבר אדפטורים בצורה יעילה, צריך ליצור קצוות נקיים וישרים - Blunt ends.
לשם כך משתמשים באנזימים:
- נוקלאזות שמסירות קצוות עודפים.
- פולימראזות שמשלימות קצוות חסרים, כאשר יש להן template.
אין צורך להיכנס לשמות הספציפיים של הפולימראזות. מה שחשוב הוא העיקרון: קודם יוצרים קצה נקי וישר, ואז מוסיפים A כדי לחבר אדפטור.
A-tailing ו־Adapter ligation
אחרי שיש קצה ישר, מוסיפים לקצה של כל מקטע A אחד. האדפטורים עצמם מכילים T-overhang, ולכן ה־A של המקטע מתאים ל־T של האדפטור כמו לגו.
לא משנה מה הרצף הגנומי של המקטע המקורי; כל המקטעים מקבלים את אותה תוספת A בקצה, ולכן ניתן לחבר אליהם אדפטורים אוניברסליים.
לאחר ההתאמה בין A ל־T, משתמשים ב־DNA ligase כדי לחבר את האדפטורים באופן יציב למקטע.
בחירת מקטעים לפי גודל (Size Selection)
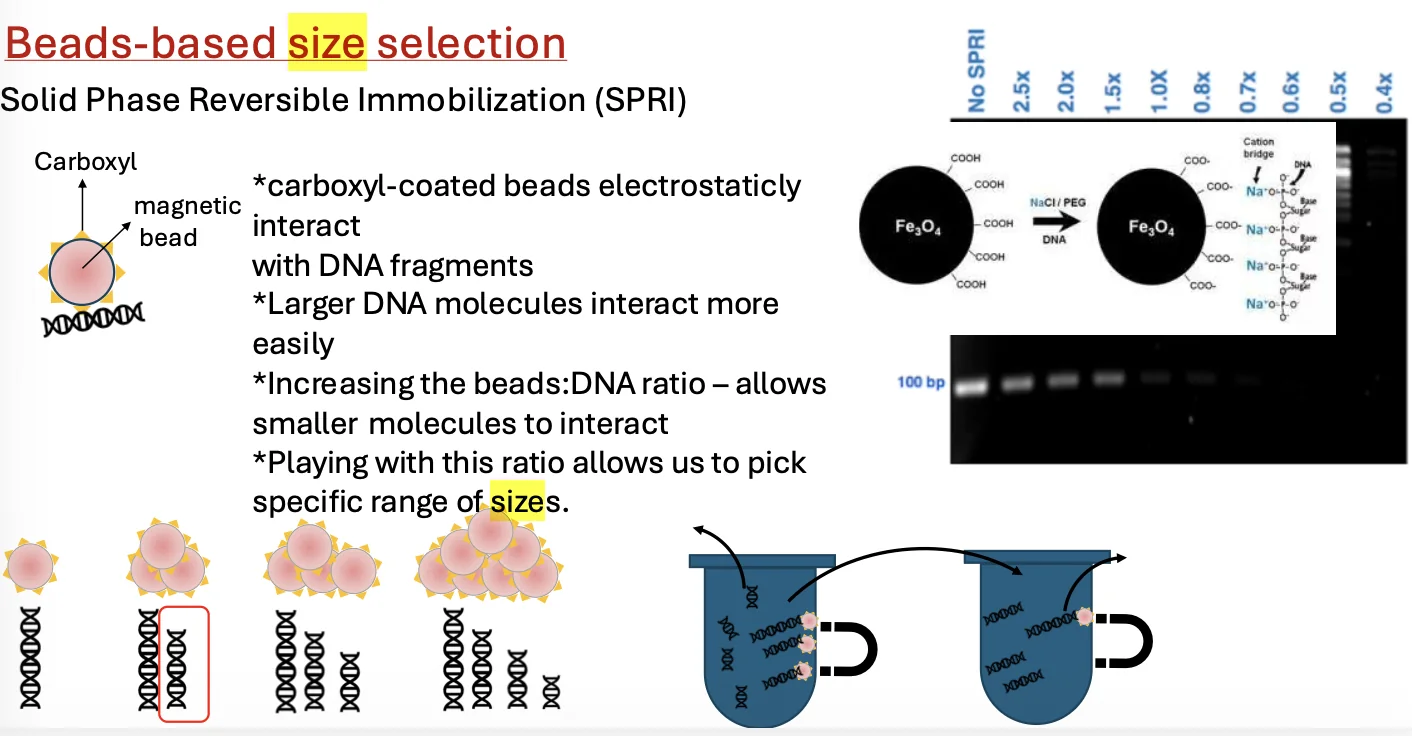
אחרי הכנת המקטעים וחיבור האדפטורים, צריך לבחור מקטעים בטווח גודל שמתאים למכונת הריצוף. הם לא חייבים להיות בדיוק באותו האורך, אבל הם צריכים להיות סביב טווח שה־sequencer יודע לרצף.
אפשר לבצע Size selection בג’ל, אבל זה פחות רצוי בספריות DNA, כי כמות החומר יקרה ומוגבלת, ובניקוי מג’ל מאבדים חומר. לכן בדרך כלל משתמשים ב־Magnetic beads.
איך משתמשים בבידים מגנטיים ל־Size selection?
בידים מגנטיים הם חרוזים קטנים שיש להם שתי תכונות:
- הם מגנטיים, ולכן אפשר למשוך אותם בעזרת מגנט.
- הם מצופים במולקולה שמקנה להם ספציפיות מסוימת.
ב־Size selection, הבידים מצופים לרוב במולקולות עם קבוצות קרבוקסיל, שיוצרות אינטראקציות אלקטרוסטטיות עם DNA.
DNA הוא מולקולה שלילית בגלל קבוצות הפוספט בשלד. ככל שמקטע DNA ארוך יותר, יש לו יותר מטען שלילי, ולכן קל לו יותר להיקשר לבידים. לכן:
- מקטעים גדולים נקשרים לבידים בקלות רבה יותר.
- ככל שמוסיפים יותר בידים, גם מקטעים קטנים יותר יכולים להיקשר.
- אם מוסיפים מספיק בידים, בסוף כמעט כל ה־DNA ייקשר.
בחירת טווח אמצעי
אם רוצים להישאר רק עם מקטעים בגודל בינוני, אי אפשר לעשות זאת בצעד אחד פשוט. לכן עושים את התהליך באיטרציות:
- בשלב ראשון נפטרים מהמקטעים הקטנים מדי.
- לאחר מכן משתמשים בתנאים אחרים כדי להפריד גם את המקטעים הגדולים מדי.
- בסוף נשארים עם טווח הגדלים הרצוי.
הריכוז והיחס בין הבידים לדגימה קובעים אילו מקטעים ייקשרו ואילו יישארו בתמיסה.
עיקרון חוזר: הביד עצמו הוא רק כלי. הספציפיות נקבעת לפי מה שמצפה אותו. ב־Size selection הספציפיות היא לגודל; ב־ChIP-seq הספציפיות תהיה לחלבון, באמצעות נוגדן.
PCR amplification ובעיות הטיה
לעיתים צריך להגביר את הספרייה ב־PCR כדי שיהיה מספיק חומר. אבל יש כאן פשרה: רוצים מספיק DNA לריצוף, אבל רוצים גם שהאוכלוסייה המוגברת תייצג נאמנה את האוכלוסייה המקורית.
PCR עלול ליצור הטייה (bias) - למשל כתלות ב־GC content או ביעילות annealing של פריימרים למקטעים שונים. לכן העיקרון הוא להשתמש במינימום מחזורי PCR שנדרש, ולא להגביר מעבר לכך.
סוגי ספריות והשלב שבו הן שונות זו מזו
אחרי שמבינים את שלבי ההכנה הטכניים, קל יותר להבין את ההבדל בין סוגי הספריות: ההבדל המרכזי הוא בדרך שבה עושים סלקציה למקטעים שרוצים לרצף.
- ב־Whole genome sequencing אין סלקציה ספציפית - רוצים את כל הגנום.
- ב־ChIP-seq הסלקציה היא לפי קישור לחלבון או למודיפיקציית היסטון.
- ב־RNA-seq הסלקציה מתחילה בזה שהמקור הוא RNA ולא DNA.
- ב־ATAC-seq הסלקציה היא לאזורים פתוחים בכרומטין.
- בשיטות מיפוי מתילציה הסלקציה/הקריאה מכוונת לדפוסי מתילציה.
- ב־Hi-C המידע הוא על קרבה מרחבית ולא רק על רצף ליניארי.
אם מבינים את עקרון הסלקציה, מבינים את רוב ההבדל בין ספריות.
ChIP-seq: זיהוי אתרי קישור של חלבונים בגנום
ChIP-seq הוא קיצור של: Chromatin Immunoprecipitation followed by sequencing, כלומר: שיקוע כרומטין בעזרת נוגדן, ולאחר מכן ריצוף.
מתי משתמשים ב־ChIP-seq?
משתמשים ב־ChIP-seq כשרוצים למפות לאורך הגנום:
- איפה חלבון מסוים נקשר ל־DNA
- איפה נמצאות מודיפיקציות מסוימות של היסטונים
- איך משתנה פרופיל הקישור בין תנאים שונים, למשל בין תא נורמלי לתא סרטני
זה כלי מרכזי באפיגנטיקה, משום שאפיגנטיקה עוסקת ברגולציה של פעילות גנים שאינה נובעת משינוי ברצף ה־DNA עצמו.
מה מקבלים בסוף?
התוצר של ChIP-seq הוא פרופיל לאורך הגנום. כאשר הרבה קריאות (reads) מגיעות מאזור מסוים, מתקבל שם פיק.
המשמעות של פיק גבוה: באחוז גבוה יותר מהתאים בדגימה, הפקטור שנחקר היה קשור לאזור הזה. לכן מספר הקריאות באזור מסוים משקף את מידת ההעשרה של אותו אזור בספרייה.
דוגמאות למיפוי פונקציונלי
ChIP-seq יכול לשמש למיפוי פונקציונלי של הגנום. למשל:
-
H3K4me3 - מודיפיקציית היסטון שמועשרת באזורי פרומוטור ובתחילת שעתוק. מיפוי שלה יכול לעזור לזהות אזורי התחלת שעתוק ולהעריך פעילות שעתוק.
-
מודיפיקציות אחרות, למשל כאלה הקשורות לאזורים סגורים בגנום, יכולות לסמן אזורים מושתקים או פחות פעילים.
לא נכנסנו לכל רשימת הסמנים; העיקר כאן הוא להבין את העיקרון: ממפים פקטור או סימן אפיגנטי כדי ללמוד איפה בגנום הוא נמצא, ומה זה אומר על פעילות האזור.
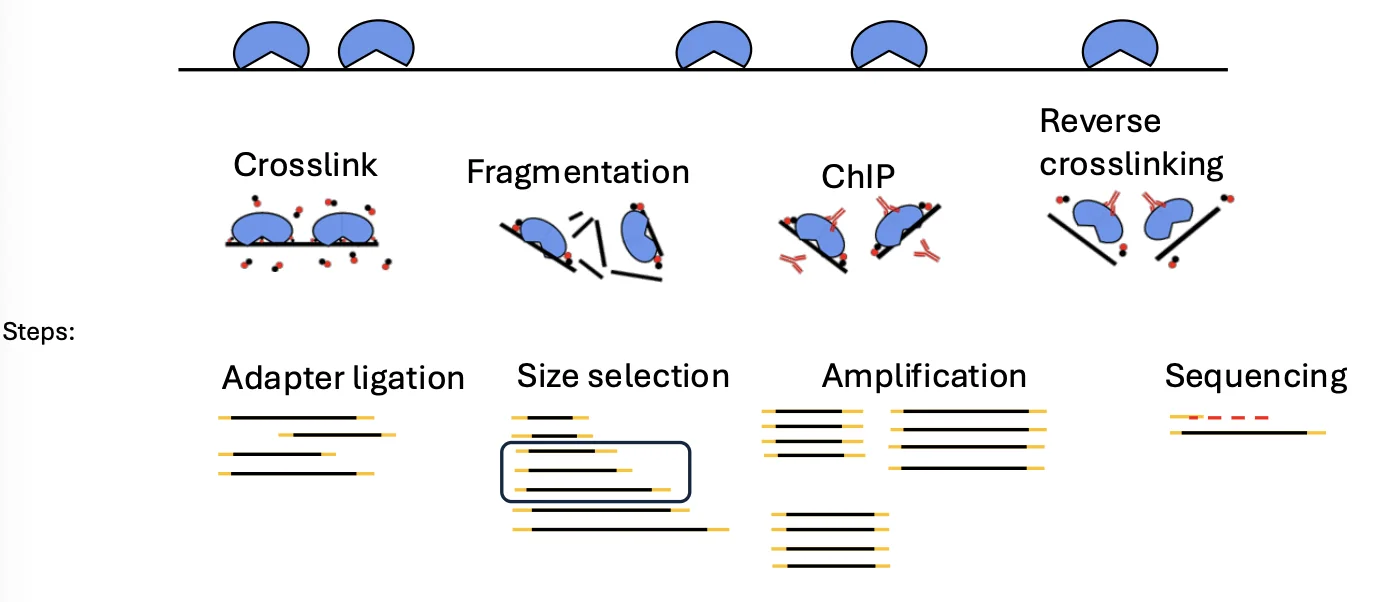
שלבי ChIP-seq
1. Cross-linking
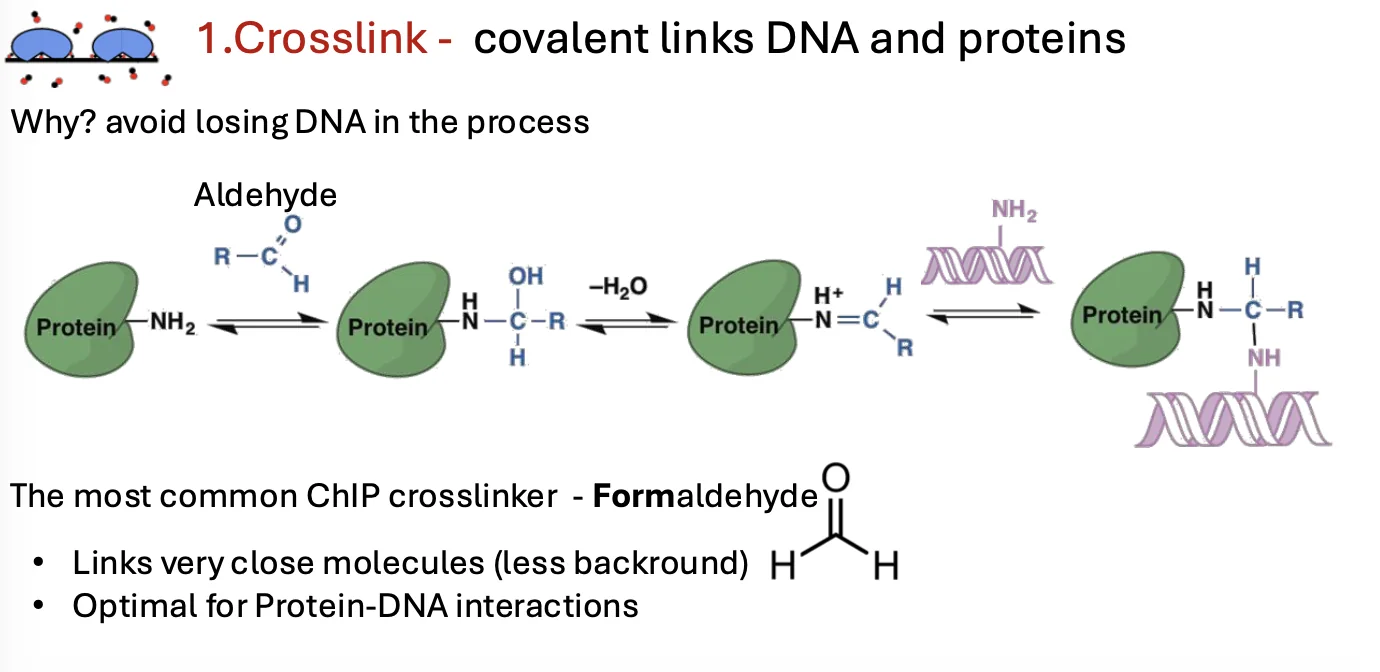
בשלב הראשון מקבעים את הקשרים בין ה־DNA לבין החלבונים הקשורים אליו. עושים זאת באמצעות מולקולות ממשפחת האלדהידים, בדרך כלל פורמאלדהיד (formaldehyde).
המטרה היא ליצור קשר קוולנטי בין ה־DNA לבין החלבון, כדי שהחלבון לא ייפרד מה־DNA בשלבים הבאים, בהם הדגימה עוברת חיתוך, שטיפות ומניפולציות נוספות.
2. פרגמנטציה (Fragmentation)
לאחר הקיבוע חותכים את ה־DNA למקטעים, למשל באמצעות סוניקציה. בשלב הזה רוב המקטעים בדגימה אינם מעניינים אותנו: חלקם לא קשורים לשום חלבון, וחלקם קשורים לחלבונים אחרים שאנו לא מעוניינים למפות.
לכן צריך שלב סלקציה.
3. Immunoprecipitation
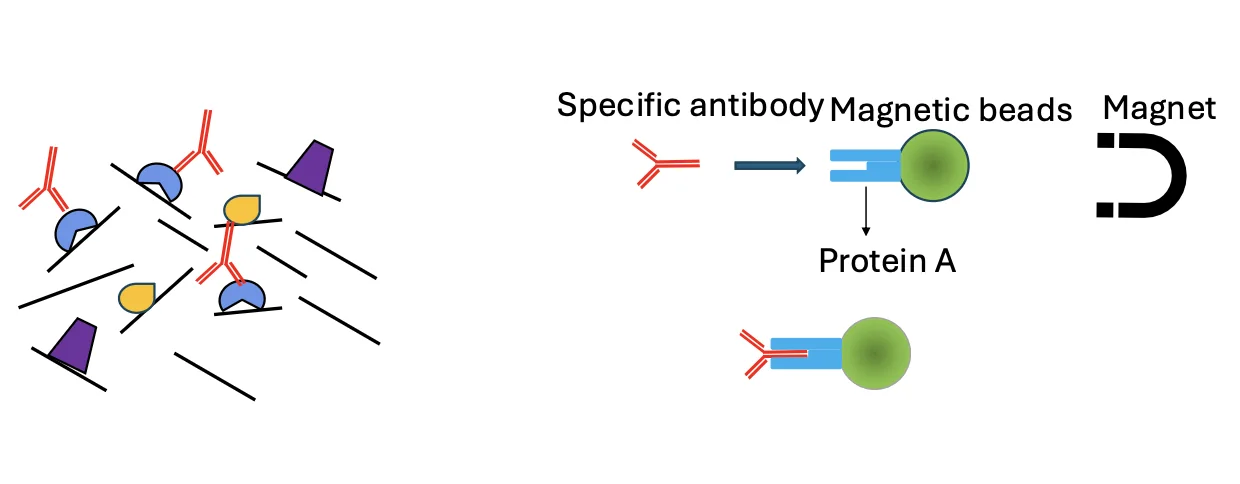
זה הלב של ChIP-seq.
כדי לדוג רק את המקטעים שקשורים לחלבון המטרה, משתמשים בשלושה מרכיבים:
- נוגדן - ספציפי לחלבון המטרה
- ביד מגנטי - כדי שניתן יהיה למשוך את הקומפלקס במגנט
- Protein A - חלבון שנקשר לאזור הקבוע של הנוגדן
הספציפיות לחלבון המטרה מגיעה מהנוגדן. Protein A ״לא יודע״ מהו חלבון המטרה; הוא פשוט נקשר לחלק הקבוע של הנוגדן. לכן, כאשר מחברים נוגדן ספציפי לביד דרך Protein A, מקבלים קומפלקס שמסוגל לדוג את חלבון המטרה ואת מקטע ה־DNA שהיה קשור אליו.
לאחר מכן מפעילים מגנט, שוטפים את כל מה שלא נקשר, ונשארים רק עם המקטעים הרצויים.
4. Reverse cross-linking
לאחר שהמקטעים הרצויים עברו העשרה, מפרקים את הקשר בין ה־DNA לחלבון. אנחנו רוצים בסוף לרצף DNA, לא חלבון.
5. המשך הכנת הספרייה
מכאן ממשיכים כמו בספריות DNA אחרות:
- תיקון קצוות
- חיבור אדפטורים
- Size selection
- אמפליפיקציה לפי הצורך
- ריצוף
- אנליזה חישובית - בעיקר Peak calling (ראו בשיעור הבא).
סיכום
ספריית DNA היא אוכלוסיית מקטעים שהוכנו לריצוף. מה שמגדיר את הספרייה הוא לא רק הטכניקה, אלא בעיקר השאלה הביולוגית: האם רוצים את כל הגנום, את האקסום, את הטרנסקריפטום, את אזורי הקישור של חלבון מסוים, או את המבנה המרחבי של הגנום.
המהפכה של NGS נובעת משני עקרונות: ריצוף מקבילי של מיליוני מקטעים ו־sequencing by synthesis, כלומר קריאת הרצף תוך כדי הסינתזה. כדי שזה יעבוד, צריך להכין ספרייה: לשבור DNA למקטעים, לתקן קצוות, לחבר אדפטורים, לבחור טווח גדלים מתאים ולעיתים לבצע אמפליפיקציה.
ChIP-seq מדגים את העיקרון הרחב של ספריות DNA: בשלב הראשון עושים סלקציה למקטעים שמעניינים אותנו - במקרה הזה בעזרת נוגדן נגד חלבון או מודיפיקציית היסטון - ואחר כך ממשיכים בשלבים הטכניים המשותפים לריצוף.
דור פסקל